

| The Peoples Republic of ChinaMedical Device Registration Certificate (In-vitro Diagnostic Reagent)(Format)Review and Approval Department:Date of Approval: (Year) (Month) (Date) |
Attachment 1
The People's Republic of China
Medical Device Registration Certificate (In-vitro Diagnostic Reagent)
(Format)
Registration certificate number:
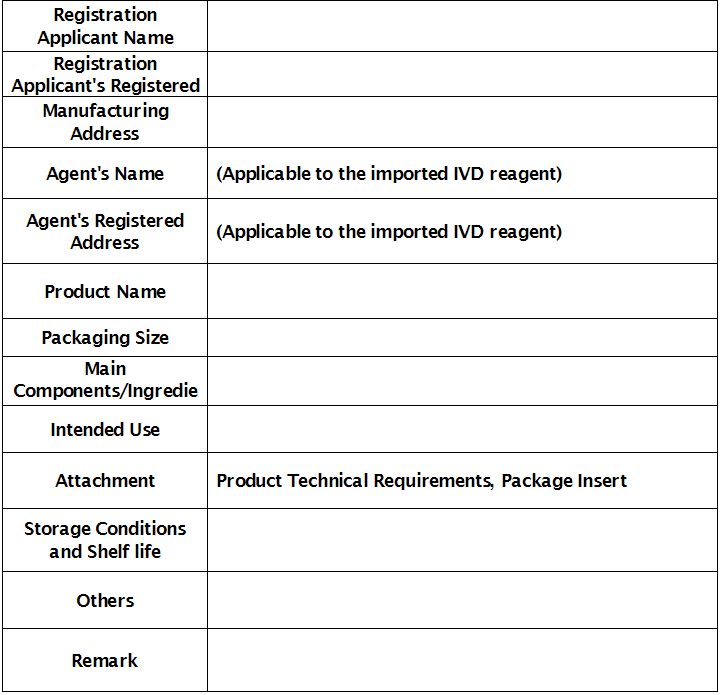
Review and Approval Department:
Date of Approval: (Year) (Month) (Date)
Expiration Date: (Year) (Month) (Date)
(Seal of the review and approval department)
Attachment 2
The People's Republic of China
Medical Device Registration Change File (In-vitro Diagnostic Reagent)
(Format)
Registration certificate number:

Examination and Approval Department:
Date of Approval: (Year) (Month) (Date)
(Seal of the examination and approval department)
Attachment 3
Requirements and Instructions for the Registration Application Documents of IVD Reagent
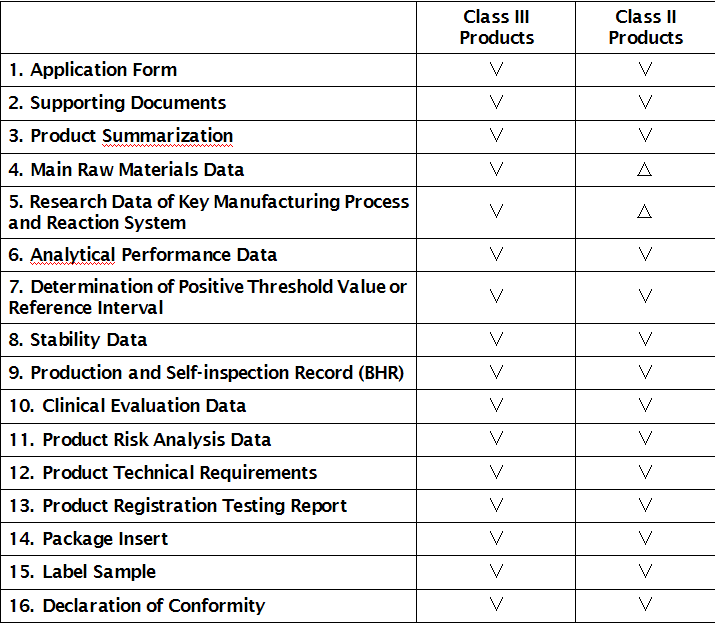
Note: Applicant shall submit the application documents according the above list and the class of product.
��: Required
��: Not necessary for application, to be kept by applicant and be provided when required by technical review.
I. Application Form
II. Supporting Documents
(I) Domestic applicant needs to provide: a copy of Business License of legal entity and a copy of Organization Code Certificate.
(II) Overseas applicant needs to provide:
1. Marketing clearance of the product issued by the medical device competent authority of the country (region) where the overseas applicant is registered or where the manufacturing site is located, or the qualification documents to legally manufacture the proposed product. If such certificates include the product category description, the category shall cover the proposed product.
2. For products not listed and regulated as medical devices in the country (region) where the overseas applicant is registered or the manufacturing site is located, the applicant shall provide the qualification documents including the marketing clearance issued by the registration/manufacturing site's country (region).
3. Qualification documents proving the applicant's compliance with the medical device Quality Management System or other quality management system in the country (region) where the application is registered or where the manufacturing site is located.
4. Authorization letter for the agent in China, Commitment letter of the agent, and a copy of Business License of the agent or a copy of Organization Code Certificate of the agent.
III. Product Summarization
(I) Intended Use of Product. Describe the intended use of the product, and background conditions of clinical indications related to intended use, such as rate of occurrence of clinical indications, populations at risk etc., and relevant clinical or laboratory diagnostic methods etc.
(II) Product Description. Describe the technique principles of product, sources and preparation methods of main raw materials, key production process, preparation and traceability (reference) of the control product and calibration product.
(III) Descriptions of Biological Safety. As the main raw materials of IVD reagent might be made from animal, pathogens, tissues and body fluid of human origin etc. or with addition of other substances, materials of human origin shall come with instructions for pathogen detection for relevant infectious disease (HIV, HBV, HCV etc.). Materials of other animal or microbiology origin shall come with relevant statement, to demonstrate safety with regards to the users and environment during transportation and application, with description of inactivation methods etc. for the above raw materials.
(IV) Summarization and evaluation of main research results related to the product.
(X) Others. The marketing clearance of similar products in China and in other countries; technical methods adopted by and clinical application of relevant products, comparison of the product against domestic or oversea predicate. For a new IVD reagent, literatures proving the relation between subjects and anticipated clinical indications shall be provided.
IV. Main Raw Materials Data
Main raw material Data shall include selection and preparation of key reaction component, quality control product and calibration product, and research data for quality standard, quantitative experiment data for quality control product and calibration product, traceability documents for the calibration product etc.
V. Research Data of Key Manufacturing Processes and Reaction System
Key production process includes: Preparation, packaging and freeze-dry of working fluid, packaging and assembly of solid phase carrier, description of derivation of color/lighting system etc. Reaction system includes acquisition and processing of samples, sample size, reagent dose, reaction conditions, calibration methods (if any), quality control methods etc.
VI. Evaluation Data of Analytical Performance
(I) The evaluation for analytical performance for IVD reagent mainly includes precision, accuracy, sensitivity, specificity, linear range or measurement range etc. Multiple batches shall be evaluated for performance; the results shall be analyzed statistically, to effectively control the production process and stability of the product quality.
If the product applies to different types of instrument, the testing data and evaluation summary on each instrument shall be provided.
If the registration application includes different packaging specifications, analysis or verification for the differences among all packaging specifications shall be provided. If performance differs among different specifications, experiment data and summary for carrying out above items using the different packaging specifications shall be submitted. If no difference in performance exists among the specifications, detailed statement for no existence of difference shall be provided with the description of the difference among all packaging specifications and potential impact.
(II) Complete traceability documents shall be submitted for calibration product.
(III) Quantitative data run on all applicable instruments shall be submitted for quality control product.
VII. Determination of Positive Judgment Value or Reference Interval
The determination and basis for the positive threshold value or reference interval shall be described in detail include the origin of the sample adopted for determination of positive threshold value or reference interval. Detailed experiment data and summary for the determination of positive threshold value and reference interval shall be provided.
Determination of positive threshold value or reference interval is not required for calibration product and quality control product.
VIII. Stability Data
It shall include real-time stability data for at least three batches of samples kept under actual storage conditions until after product shelf life, with sufficient considerations for unfavorable conditions during storage, transportation and application for carrying out the relevant stability study. Evidence and specific test methods and processes for the study methods for stability shall be described in detail.
IX. Production and Self-inspection Record
Copies of production and self-inspection records of three consecutive batches shall be provided.
X. Clinical Evaluation Data
(I). Clinical Trial. The applicant shall follow the applicable technical guidelines to carry out clinical trials for IVD reagents that require clinical trial. The following clinical trial data shall be provided:
1. Written Advice of Ethics Committees Agreeing the Clinical Trial.
2. Clinical Trial Protocol: signed by principal investigators in charge of the clinical trial, stamped by the clinical trial site, signed by the person in charge of statistical analysis and stamped by the statistical analysis agency, and stamped by the applicant.
3. Clinical trial report of each clinical trial site: each clinical trial report shall be stamped by the clinical trial site; the cover of the report shall include the generic name, start date of the trial, completion date of the trial, principal investigator (signature), trial site (stamp), signature of person in charge of statistical analysis and site (stamp) in charge of statistical analysis, applicant (stamp), contact of applicant and contact methods, date of report, storage location of original data for the IVD reagent used in the trial.
4. Summary report for all clinical trial results: the summary report shall be completed by the leading clinical trial site or the applicant, with the same cover content as the clinical trial report from each clinical trial site.
5. Appendix for clinical trial report: detailed data of the clinical trial, including basic information of other trial methods or other diagnostic reagents adopted during the clinical trial, such as trial methods, origin of diagnostic reagent, package insert and registration approval, all trial data of the clinical trial (shall be signed by operator and reviewer of the clinical trial, and stamped by the trial site), main reference literatures, background of principal investigator, and other matters that the applicant wish to clarify etc.
(II). For the products in the clinical trial exemption list of IVD reagent issued by CFDA, relevant clinical evaluation data shall be submitted. According to relevant guidelines (if any), the applicant shall provide assessment of clinical performance of IVD reagents through safety and effectiveness data of the product such as the evaluation of clinical sample covering the indications and interference factors, general literature, and historical clinical data etc. and source information for the clinical sample used.
(III). Clinical trial data completed abroad or summary report of clinical application shall be submitted for imported product.
(IV). Clinical trial data is not required for calibration product and quality control product.
(V). The stamp of the clinical trial site in this section refers to the organization stamp of the clinical trial site.
XI. Product Risk Analysis Data
The risk control report shall be based on the risk analysis, risk evaluation and corresponding risk control based on indications, possible operational error, features associated with safety, determination of known and foreseeable hazard etc. and evaluation of risk to the patients. It shall meet relevant requirements of industrial standards.
XII. Product Technical Requirements
Provided that the quality of the raw material and production process are stable, the applicant shall compile the Product Technical Requirements according to the product development, early stage clinical evaluation etc., national standards, industrial standards and relevant literature, and Guidelines for Establishment of Technical Requirements for Medical Devices, the content shall mainly include product performance index and test methods. Product Technical Requirements for Class III IVD reagents shall clearly specify requirements for main raw materials, production technological process and semi-finished products in the form of appendices.
The technical requirements for imported products shall include both English and Chinese versions. The English version shall be signed and notarized by the applicant; the Chinese version shall be signed and stamped by the applicant or the agent. The Product Technical Requirements in Chinese shall be provided in duplicate, together with a declaration that the two copies are identical to each other.
XIII. Product Registration Test Report
The Registration Test Report and Product Technical Requirements Pre-evaluation Comments provided by a qualified medical device testing institution shall be provided. Where national standard/reference product is available, the national standard/reference product shall be used for registration examination, and shall meet relevant requirements.
XIV. Package Insert
For domestic product, the applicant shall establish the package insert according to relevant requirements of Guidelines for Developing Package Insert for IVD Reagents and relevant technical guidelines.
For imported product, the applicant shall submit the original package insert approved or certified by foreign authority and Chinese translations. The agent shall establish the package insert according to relevant requirements of Guidelines for Developing Package Insert for IVD Reagents and relevant technical guidelines.
Two copies of the package insert established according to the guidelines shall be submitted, together with a declaration that the two copies are identical to each other.
XV. Sample label
The requirements of Administrative Provisions on the Package Insert and Labels for Medical Devices shall be met.
The label on the product external packaging shall include the generic name of the product, applicant��s name, manufacturing address, product batch number, precaution, storage conditions and shelf life etc.
The label shall indicate the Chinese name and batch number of the components in IVD reagents such as calibration product, quality control product and cleaning liquid etc. If components of different batch number are not interchangeable within the same batch of product, the product batch number as well as the component batch number shall be indicated.
Labels approved or certified by foreign authority and the Chinese translation shall be submitted for imported product shall, the Chinese sample label shall be submitted according to the above requirements.
XVI. Declaration of Conformity
(I). The applicant shall declare that the product meets relevant regulations and requirements of Administrative Measures on In-vitro Diagnostic Reagent Registration, that the classification of the product is in line with Administrative Measures on In-vitro Diagnostic Reagent Registration and IVD Reagent Classification sub-catalog, and that the product meets the current national standards and industrial standards with a list of standards met.
(II). The applicant shall declare that all of the materials submitted are authentic (which shall be issued by the registration applicant for domestic product, or shall be issued by the registration applicant and its agent for imported products).
XVII. Other
Item III, IV, V, VI, VII, VIII, IX, and XI in the list of application documents shall be submitted by the applicant for imported product.
Attachment 4
Requirements and Instructions for Extension Registration Application Documents for IVD Reagent
I. Application Form
II. Supporting Documents
A domestic applicant needs to provide a copy of business license of legal entity and a copy of Organization Code Certificate. An overseas applicant needs to provide the authorization letter for the agent in China, commitment letter of the agent, and a copy of business license of the agent or a copy of Organization Code Certificate of the agent.
Note: No marketing clearance, issued by the country (region) where the application is registered or where the manufacturing site is located, is required when applying for extension registration for an imported medical device.
III. Declaration letter for no product change
The registration applicant shall provide a declaration letter for no change to the product.
IV. A copy of the original medical device registration certificate (and appendices), and copies of each approval of changes in medical device registration
V. Product analysis report during the validity period of the registration certificate
(I). The product's clinical application, customer complaints, and actions taken.
(II). Medical device adverse event summary analysis and evaluation report, in which all of the suspected adverse events occurred after the product is placed on the market shall be listed and the solutions taken by the manufacturer in each situation shall be explained. The above-mentioned adverse events shall be analyzed and evaluated, the root causes of the adverse events shall be illustrated, and the impact on the product's safety and effectiveness shall be explained.
(III). The sales status of the product in all countries and regions.
(IV). The results of the supervision and random inspection of the product, if any.
(V). If any product recall occurred, the root cause, process and results of the recall shall be explained.
(VI). If the original medical device registration certificate specified any work to be continued, a relevant summary report shall be provided, with corresponding materials being attached.
VI. Product testing report
If the mandatory standard for the medical device has been revised, a product testing report showing the product meeting the new requirements shall be provided. The product testing report can be a self-testing report, an entrusted testing report, or a testing report showing the product compliance with the provisions defined in the notice on executing the mandatory standard. Of that, the entrusted testing report shall be issued by a qualified medical device testing institution.
If there is a release or update of the national standard product or the reference product, the testing report demonstrating that the product can meet the requirements of the national standard product or reference product shall be provided. The product testing report can be a self-testing report, an entrusted testing report, or a testing report showing the product's compliance with the applicable notice provisions.
VII. Declaration of Conformity
(I). The registration applicant shall declare that the product meets the relevant requirements of Administrative Measures on IVD Reagent Registration, and declare that the product meets the current national standards and industrial standards and provide the list of standards being met.
(II). The applicant shall declare that all of the materials submitted are authentic (which shall be issued by the registration applicant for domestic product, or shall be issued by the registration applicant and its agent for imported products).
VIII. Other
If any change to the package insert and/or product technical requirements is made during the validity period of original registration certificate, the package insert and/or product technical requirements, updated in accordance with the Registration Revision Document, shall be provided respectively in duplicate.
Attachment 5
Requirements and Instructions for the Change Registration Application Documents for IVD Reagent
Requirements and Instructions for Application Documents of Registered Items Change
I. Application Form
II. Supporting Documents
(I). A domestic applicant needs to provide:
1. A copy of Business License of legal entity.
2. A copy of Organization Code Certificate.
(II). An overseas applicant needs to provide:
1. Applicable documents if the change requires marketing clearance issued by the medical device competent authority of the country (region) where the overseas application is registered or where the manufacturing site is located, or requires the certifying documents of the business entity; or an explanation if the change requires no approval by the medical device competent authority of the country (region) where the application is registered or where the manufacturing site is located.
2. Authorization letter for the agent in China, commitment letter of the agent, and a copy of Business License of the agent or a copy of Organization Code Certificate of the agent.
III. Declaration Letter from the applicant for the changes
IV. A copy of the original medical device registration certificate (and appendices), and copies of each approval of changes in medical device registration
V. Requirements for change-related application documents
(I). Name change of registration applicant:
Notice on Business Name Change Approval (for domestic applicant) and/or description on the changes as well as supporting documents.
(II). Address change of registration applicant:
Detailed description on the changes as well as supporting documents.
(III). Manufacturing addresses change of domestic IVD reagent:
The updated manufacturing license after changes.
(IV). Agent change:
1. Registration applicant��s declaration on the change of agent.
2. Authorization letter for the new agent, and commitment letter of the new agent.
3. A copy of business license or a copy of Organization Code Certificate after the agent is changed.
(V). Agent��s address change:
Copies of Business License or copies of Organization Code Certificate before and after change.
VI. Declaration of Conformity
(I). The registration applicant shall declare that the product meets the relevant requirements of Administrative Measures on IVD Reagent Registration, and declare that the product meets the current national standards and industrial standards and provide the list of standards being met.
(II). The applicant shall declare that all of the materials submitted are authentic (which shall be issued by the registration applicant for domestic product, or shall be issued by the registration applicant and its agent for imported products).
Requirements and Instructions for Change Application Documents for Permitted Items
I. Application Form
II. Supporting Documents
(I). A domestic applicant needs to provide:
1. A copy of Business License of legal entity.
2. A copy of Organization Code Certificate.
(II). An overseas applicant needs to provide:
1. Applicable documents if the change requires marketing clearance issued by the medical device competent authority of the country (region) where the overseas application is registered or where the manufacturing site is located permitting the free sales of the product, or requires the certifying documents of the business entity; or an explanation if the change requires no approval by the medical device competent authority of the country (region) where the application is registered or where the manufacturing site is located.
2. Authorization letter for the agent in China, the agent's letter of commitment, and a copy of business license of the agent or a copy of Organization Code Certificate of the agent.
III. Declaration letter of the applicant for the changes
(I). Reason and purpose for change.
(II). Technical analysis of the possible impact of change on the performance of the product.
(III). Product risk analysis data relating to product change.
IV. A copy of the original medical device registration certificate (and appendices), and copies of each approval of changes in medical device registration
V. Other technical data of specific change
(I). The following documents shall be submitted when the suppliers of main materials such as antigens, antibodies, etc. are changed:
1. Research data about the change of antigen/antibody materials;
2. Evaluation data of analytical performance.
3. Clinical trial data.
4. Product Technical Requirements and Package Inserts before and after change.
(II). The following documents shall be submitted when test conditions, positive threshold value or reference interval is changed:
1. Detailed experiment data used to determine the changed test conditions and the changed reference values (or reference range).
2. Clinical trial data.
3. Product Technical Requirements and Package Inserts before and after change.
(III). The following documents shall be submitted when storage conditions and/or shelf life is changed:
1. Experiment data of product stability study.
2. Product Technical Requirements, Package Inserts and sample label before and after change.
(IV). The following documents shall be submitted when Product Technical Requirements are changed without degrading the effectiveness of product:
1. Evaluation data of analytical performance.
2. Product Technical Requirements and Package Inserts before and after change.
(V). The following documents shall be submitted when manufacturing addresses of imported IVD reagents are changed:
1. Quality system evaluation reports of manufacturing addresses change of imported IVD reagents, if any.
2. Certifying documents proving the new manufacturing site's compliance with the medical device quality management system or other quality management system effective in the country (region) where the manufacturing site is located.
3. Experiment data of analytical performance evaluation if products utilizing new manufacturing site.
4. Product Technical Requirements and label sample after change.
(VI). The following documents shall be submitted when the wording in the Package Inserts and/or Product Technical Requirements are changed without changing any technical contents:
1. The change description of Package Inserts and/or Product Technical Requirements shall be submitted, with a comparison sheet of changes.
2. Package Inserts and/or Product Technical Requirements before and after change.
(VII). The following documents shall be submitted when packaging specification is changed:
1. Product Technical Requirements, Package Inserts and label sample before and after change, if involving.
2. Determine whether there is a difference in performance between the specifications of changed packaging and that of marketed packaging. If performance differs among different packaging specifications, experiment data and summary for carrying out above items using the different packaging specifications shall be submitted. If no difference in performance exists among the different packaging specifications, detailed statement for no existence of difference among different packaging specifications shall be provided, to elaborate in particular the difference of the packaging specifications and possible impact.
(VIII). The following documents shall be submitted when applicable instruments are changed:
1. Testing data of analytical performance evaluation when a new applicable instrument is utilized.
2. Product Technical Requirements, Package Inserts and label sample before and after change (if involving).
(IX). The following documents shall be submitted when adding clinical indications:
1. Testing data of analytical performance evaluation when a new clinical indication is added (if applicable).
2. Clinical trial data of the added clinical indication.
3. Product Technical Requirements and Package Inserts before and after change.
(X). The following documents shall be submitted when sample types for clinical application are added:
1. For clinical trial data adopting comparison of additional clinical trial sample types against approved sample types, clinical trial comparing marketed predicate product comparable to the sample type can be selected, if the additional sample types and the original approved sample types are not directly comparable.
2. Product Technical Requirements and Package Inserts before and after change.
(XI). Relevant test data shall be submitted according to the change when those changes might affect the effectiveness of products.
(XII). Testing data for verification of possible impact of the change on the product shall be submitted according to the specific change (if involving).
VI. Declaration of Conformity
(I). The registration applicant shall declare that the product meets the relevant requirements of Administrative Measures on IVD Reagent Registration, and declare that the product meets the current national standards and industrial standards and provide the list of standards met.
(II). The applicant shall declare that all of the materials submitted are authentic (which shall be issued by the registration applicant for domestic product, or shall be issued by the registration applicant and its agent for imported products).