

| Approved by (Department):Date of Approval: YYYY MM DDDate of Expiry: YYYY MM DD(Seal of Evaluation and Approval Department)Appendix 2Permission of change to registration for me |
Appendix 1
Registration Certificate for Medical Device of People's Republic of China
(Format)
Registration Certificate No:
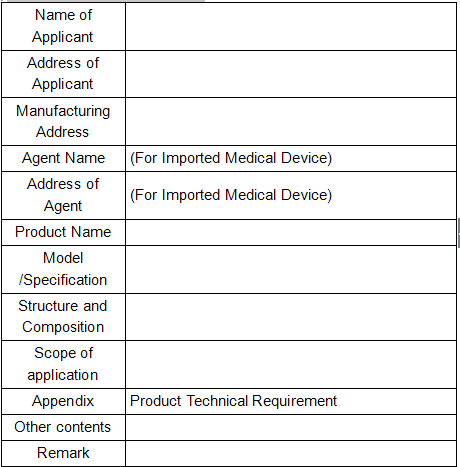
Approved by (Department): Date of Approval: YYYY/MM/DD
Date of Expiry: YYYY/MM/DD
(Seal of Evaluation and Approval Department)
Appendix 2
Permission of change to registration for medical device of People's Republic of China
(Format)
Registration No.��

Approved by (Department): Date of Approval: YYYY/MM/DD
(Seal of Evaluation and Approval Department)
Appendix 3
CFDA Approval for Medical Device Clinical Trial
(Format)
Approval No.:

Approved by (Department): Date of Approval: YYYY/MM/DD
(Seal of Evaluation and Approval Department)
Appendix 4
Requirements and Instructions for Medical Device Registration Application
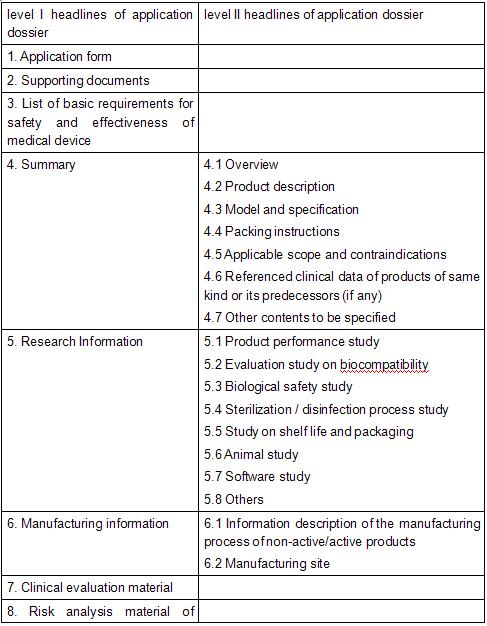
The application dossier shall have an index which covers all level I and level II headlines, and separately compile page number for the corresponding information to each level II headline.
I. Application form
II. Supporting documents
(I) Domestic applicant shall provide:
1. Copies of business license and organization code certificate
2. When applying for registration of domestic medical devices according to Special Procedure of Approval and Evaluation for Innovative Medical Devices, applicant shall provide a notice of application for reviewing ��Special procedure of approval and evaluation for innovative medical devices��, and if the sample products are produced by entrusted manufacturers, manufacturing license of the entrusted manufacturer and consignment agreement shall be provided. The scope of manufacturing license shall cover the category of the submitted products.
(II) Imported Medical Device applicant shall provide:
(1) Supporting documents of marketing authorization or certificate of the product issued by authority of the country (or region) where the applicant��s headquarter or manufacturing site is located, and the authorization/qualification documents of the enterprise.
(2) If the product is not managed as a medical device by authority of the country (or region) where the Imported medical device applicant is located, applicant shall provide relevant supporting documents, including supporting documents of marketing authorization and quantification certificate of manufacturer issued by authority of the country (or region) where the registration office or manufacturing site is located.
(3) Power of attorney of the foreign applicant for designating agent in China, copies of the letter of commitment and business license or copy of organization registration certificate of agent.
III. List of basic requirements for safety and effectiveness of medical device
The list is a document describing that the product complies with methods used in all applicable requirements of the List for Basic Requirements for Safety and Effectiveness of Medical Device (see Appendix 8) and proving its conformity. The reasons shall be given for all inapplicable requirements in the List for Basic Requirements for Safety and Effectiveness of Medical Device.
For documents included in application dossier, their specific locations in the dossier shall be provided; for those not included in application dossier, the name of documentary evidence and its document number in QMS shall be noted.
IV. Summary
(I) Overview
Describe the management category, criteria for determining the classification code and device name of the product
(II) Product description
1.Non-active medical devices
Describe the product��s operation principle, mechanism of operation (if applicable), structure & composition (including supporting accessories), main raw materials and different features with other products of same kind; and graphic or charts with explanation shall be provided as necessary.
2. Active medical devices
Describe the product��s operation principle, mechanism of operation (if applicable), structure & composition (including supporting accessories), main function and functions of componential parts (critical components and software) and different features with other products of same kind; and graphic or charts with explanation shall be provided as necessary.
(III) Model and specification
For the products with various models and specifications, a detailed list shall be made for differences between each model and specification from the perspective of structure or composition (or configuration), function, product feature, operating mode, performance indices and others. Comparison table, graphic or charts with explanation shall be used to summarize the difference.
4. Packing instruction
Product packaging information and packing instruction of accessories sold together with such products; for sterilized device, original packaging information corresponding to sterilization method shall be indicated.
(V) Intended use and contraindication
(1) Intended use: Specify the treatment and diagnosis provided by the product meet the purpose defined in Article 76 of the Regulations for the Supervision and Administration of Medical Devices, and describe the applicable treatment periods of the product (e.g., post-treatment monitoring, rehabilitation, etc.); define the target users and the skills/knowledge/trainings required by users to operate such products; Explain the operation pattern: single use or reusable; specify medical devices intended to be used in combination with the product.
(2) Intended service environment: intended use place of such products, including hospital, medical/clinical laboratory, ambulance and home, and ambient conditions that may affect its safety and/or performance (for instance, temperature, humidity, power, pressure and movement);
(3) Target population: Targeted patients information (such as adults, children or neonates), the standards information for patients to select and relevant parameters shall be monitored during use;
(4) Contraindication: if applicable, the medical device shall specify the diseases or conditions (e.g., children, elderly people, pregnant and breast-feeding women, and patients with hepatic and renal insufficiency) that they cannot be used.
(VI) The reference information of domestic and Imported medical device products of same kind(if any) or its predecessor shall be given as well as the purpose of interpretation the goal of product research and development. For products of same kind, the reason for choosing them as the research & development shall be provided as reference.
Comparisons of the product and reference (product of same kind or its predecessors) shall be given from their similarities and differences in a list in terms of operation principles, structure & composition, manufacturing materials, performance indices, operating mode (e.g. implantation, intervention), and applicable scope.
(VII) Other contents to be specified. With regard to components or accessories approved by relevant departments, approval number and copy of approval document shall be provided; products that are intended to be used in conjunction with other medical devices or universal adaptable products to work together for the desired function shall be indicated; the physical, electrical connection type between components of combination medical devices in the system shall be indicated.
V. Research Information
Provide the applicable research materials for the submitted product.
(I) Product performance study
The information shall cover research information for performance study, technical requirements study and its compiling instructions, including performance and safety indices (e.g. electric safety, electromagnetic compatibility, and radiation safety), determination basis for other QC-relevant indices, adopted standards or methods and the theoretical basis and reasons for their being adopted.
(II) Biocompatibility evaluation study
The biocompatibility evaluation shall be made on the materials that are in direct/indirect contact with the patients and users in the final product.
The information on biocompatibility evaluation shall include:
1. Basis and method of biocompatibility evaluation.
2. Description of materials used in the product and their characteristics when in contact with human body.
3. Reasons and arguments for implementation or exemption of biological test.
4. Evaluation on the available data or test results.
3. Information on bio-safety research
For the products with biological safety risks containing allogeneic materials, materials of animal origin, or biologically active substances, the biological safety research information concerning relevant materials, and biologically active substances shall be given, including the description of processing, preservation, testing and treatment process of the tissue, cell and materials acquisition. Moreover, it is needed to demonstrate source (including screening details of donors) and describe the verification testing on the methods removing or inactivating virus, other pathogens and immunogenic substances during the manufacturing process. The brief summary for process assessment shall be provided.
(IV) Information on disinfection/ sterilization process validation
1. Sterilization by manufacturer: Specify sterilization process (methods and parameters) and sterility assurance level (SAL), and provide the sterility validation report.
2. Sterilization by end user: Recommend sterilization process (methods and parameters) specified and provide the evidence. For products that can tolerate sterilization at least twice, supporting materials of product��s resistance to recommended sterilization methods shall be provided.
3. Residual toxicity: Where sterile device is provided and methods used for sterilization may generate residue easily, the adopted residue and treatment methods shall be determined and supporting materials shall be offered.
4. End user��s disinfection: it is required to specify the recommended cleaning/sterilization process (methods and parameters) and provide the evidence.
(V) Confirmation of shelf life and packaging of products
1. Confirmation of shelf life: Shall provide the report and protocol of shelf life verification.
2. For medical devices with limited re-use times, provide the validation data of use times
3. Packaging and packaging integrity: maintain packaging integrity of device within the declared shelf life under the packaging and distribution environment
(VII) Software study
For products containing software, the independent descriptive document of medical device software shall be provided. The document shall include basic information, realization process and core algorithm, the details depend on the security level and sophisticated degree of software. Provide a statement about software version naming rules, specify all fields and their meanings of software version, and determine the complete version of software and its identification version used for release.
(VIII) Other information
Other research information or literature provided the safety or effectiveness of the product.
VI. Manufacturing Information
(I) Non-Active medical device
The manufacturing process shall be clearly indicated and the key technology and special process shall be given as well as explanation of its in-process control point. The use of various processing agent and control over impurities (such as residual monomers, residues of small molecules, etc.) during the manufacturing shall be clearly indicated. Furthermore, the basis for determining the processing technologies affecting the product performance, and related research information or literature shall be available.
(II) Active medical devices
The descriptive information on the manufacturing process of the product shall be contained, for which, the form of flow chart can be used for giving an overview of its in-process control points.
Note: For some active medical devices (for example: cardiac pacemakers and leads), the description on manufacturing process information regulated in article VI.1 shall be taken.
(III) Manufacturing site
If a product is manufactured at multiple research and manufacturing sites, the actual situations of each research and manufacture site shall be given.
VII. Clinical evaluation material
Submit the clinical evaluation material according to the relevant provisions; imported medical device shall provide clinical evaluation material used in their market application with foreign medical device authorities.
VIII. Risk analysis material of products
The risk analysis material of products is formed by recording the risk management process and evaluation results of products. The traceability of each of following processes assessed as harmful shall be provided:
(I) Risk analysis: Include the assessment of applicable scope and safety-related characteristics of the medical device, the harm assessment, and the risk evaluation of each harmful situation.
(II) Risk evaluation: Evaluate and decide whether or not to reduce the risks for each assessed harmful situation.
(III) Validation reports of risk control measures and its implementation (e.g., medical electric safety, biological evaluation, etc.)
(IV) Evaluate the acceptability of one or more residual risks.
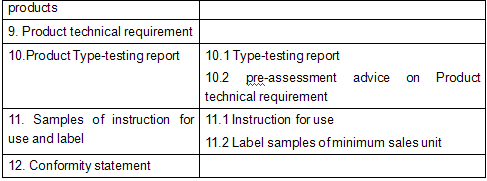
IX. Product Technical Requirement
The product technical requirement for medical device shall be compiled according to the provisions of the Compiling Guidelines of Product Technical Requirement for Medical Device. The Product technical requirement is submitted in duplicate with statement of identity of the two copies.
X. Product Type-Testing Report
Such report and pre-assessment advice shall be issued by medical device testing institutions recognized by the CFDA.
XI. Instruction for use of the product, designed package and label samples of minimum sales unit
Comply with the relevant regulatory requirements.
XII. Conformity statement
(I)The applicant declares that the product complies with the relevant requirements of the Provisions for Medical Device Registration and the relevant regulations; declares that the product complies with the classification requirements of the Medical Device Classification Rules; declares that the product complies with the current national standards, industrial standards, and provides an up-to-standard list
(II) Self-assurance statement for the authenticity of materials submitted (for domestic products, the statement is issued by the applicant; for imported products, by the applicant and the agent).