

| һ��ǰ�Խ��������Գ�����ϸ����������1 (Programmed cell death protein 1, PD-1) ��������������1 (Programmed death-ligand 1, PD-L1)���������Ƽ�Ϊ�������������ڶ������������ȡ����ͻ���� |
һ��ǰ��
���������Գ�����ϸ����������1 (Programmed cell death protein 1, PD-1)/��������������1 (Programmed death-ligand 1, PD-L1)���������Ƽ�Ϊ�������������ڶ������������ȡ����ͻ���Խ�չ��һϵ�п�����ҩ��Ӧ�����ٴ���PD-L1����Լ���ĿǰӦ����Ϊ�㷺����������������Ⱥɸѡ����ЧԤ��������־�������֯��ѧ��immunohistochemistry��IHC����ơ������黯�������������������֯PD-L1����״̬��һ����Ч�ҳ��õķ������㷺Ӧ���ڶ��ֶ��������У���ʶ�����Ԥ����ܴ����������л���Ļ��ߡ�
���������黯����PD-L1����Լ���������ͽ���ж���Ϊ���ӣ��������Լ��ٴ�Ӧ�õ�ȷ�����ظ����Dz�Ʒ��Ҫ���ٴ����ܣ���ָ��ԭ��ּ�ڹ淶��Ʒ������������ٴ��о��Ŀ�չ��
��ָ��ԭ�������PD-L1����Լ�����������ٴ��о���һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����á����ļ�Ϊ�������˺������Աʹ�õ�ָ�����ļ������漰ע������������������Ϊ����ǿ��ִ�У������ܹ����㷨��Ҫ�������������Ҳ���Բ��ã���Ӧ�ṩ��ϸ���о����ϡ���ָ��ԭ���������з��桢����ϵ����ǰ��֪ˮƽ���ƶ��ģ����ŷ��桢���IJ������ƺͿ�ѧ�����IJ��Ϸ�չ�����ļ��������Ҳ����ʱ���е�����
�������÷�Χ
��ָ��ԭ�������ڻ��������黯����PD-L1����Լ��������Ʒ���ڶ��Լ�������Ը������̶ֹ�ʯ������FFPE����������֯��PD-L1���ı���ˮƽ��
�����ٴ�����Ҫ��
PD-L1����Լ�����������ٴ��о��������������ݣ�������������Ƭһ�����о����ٴ�����Ŀ�չ���������ƶ��Լ������д�Ⱦ�Ӧ������ط��漰����������Լ��ٴ����鼼��ָ��ԭ��Ҫ��
��һ���ٴ��������
1���������飨Ring study��
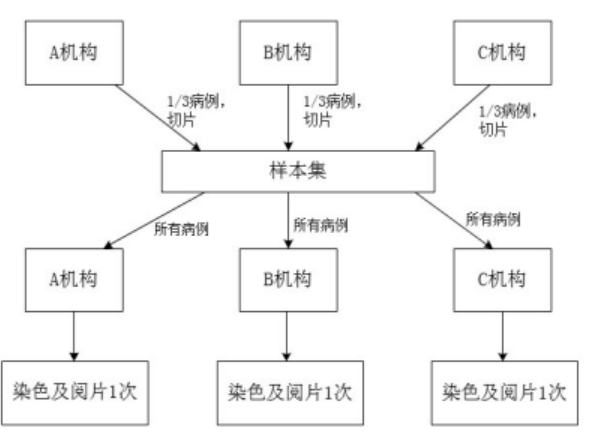
���������ǴӲ�����Ƭ��ʼ�������Լ�˵�����������������Ⱦɫ������ж������۸��ٴ����������ͬһ��������Ⱦɫ������жϵ�һ���ԡ�
��������Ӧѡ�����������ٴ����������չ����������IJ���Ϊ�����ٴ����������ͬ�ṩ��������ɵ��������������������������У�ÿ���ٴ���������ṩ�����Բ��������Բ������ܲ�����Ӧ��Ծ��⡣ÿ������Ӧ�����ṩ�㹻���ΰ����걨��Ʒ˵������м��IJ�����Ƭ������
�ٴ�����ǣͷ��λ�о��߽���������ÿһ�����IJ�����Ƭ������ÿ���ٴ������������֤ÿ���ٴ�����������ܹ��Ըò��������걨��Ʒ˵�����������һ��Ⱦɫ������������ˣ�ÿ���ٴ�����������ܶ������������в����IJ�����Ƭ����Ⱦɫ������ж���
���ٴ���������յ��ٴ�����ǣͷ��λ����IJ�����Ƭ��ѡ�����һ���������о��ߣ������걨��Ʒ˵�����Ҫ��Բ�����Ƭ����Ⱦɫ������ж������������������Ⱦɫ���龡����ɡ������ٴ��������ͨ�����������в�����Ƭ����Ⱦɫ���ж������ո������������γɻ�����������ݼ������ں���ͳ�Ʒ���������������Ƽ�ͼ1��
ͼ1.�����������ʾ��ͼ
2����Ƭһ�����о�
��Ƭһ�����о��Ķ���Ϊ���걨��Ʒ���ղ�Ʒ˵�������Ⱦɫ�IJ�����Ƭ�����ٴ���������о��߸��ݲ�Ʒ˵�����н���ж���������Ƭ�����ж������۲�ͬ�о��߶�Ⱦɫ��Ƭ�ж������һ���ԡ�
��Ƭһ�����о�Ӧѡ�����������ٴ�����������о����ݰ�����������ͬһ����ҽʦ��Ƭһ�����о��������ڲ�ͬ����ҽʦ��Ƭһ�����о�����ͬ�����䲡��ҽʦ��Ƭһ�����о������ٴ���������ɸ����ٴ��������ݣ��ڿ�ѧ��Ƶ�ǰ���½�������Ƭһ�����о��ϲ����У���Ӧ���ٴ����鷽������ȷ��������ͬ�о��߲���ľ����о����ݣ��ٴ�����Ӧ�ϸ����ٴ����鷽�����С�
2.1������ͬһ����ҽʦ��Ƭһ�����о�
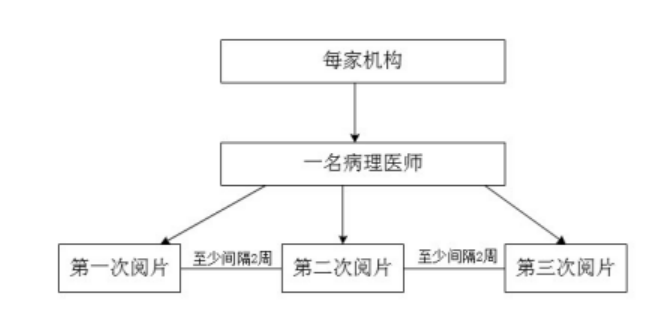
������ͬһ����ҽʦ��Ƭһ�����о�Ӧ�������ٴ����������չ��ÿ���ٴ����������Ա��������ṩ���ٴ���������������Ⱦɫ�IJ�����Ƭ���б��о��������ٴ�����IJ�����ƬӦ�����ж����Ϊ���Ե���Ƭ���ж����Ϊ���Ե���Ƭ��
ÿ���ٴ��������ѡ��һ��һ�������IJ���ҽʦ�Ըû�����������в�����Ƭ����3���ж���ÿ���ж����һ����ʱ�䣬ͬһ����ҽʦ�IJ�ͬ����Ƭ�����ɷ������ݼ���������ͬһ����ҽʦ��Ƭһ�������������ͼ2��
ͼ2.������ͬһ����ҽʦ��Ƭһ�����������ʾ��ͼ
2.2�����ڲ�ͬ����ҽʦ��Ƭһ�����о�
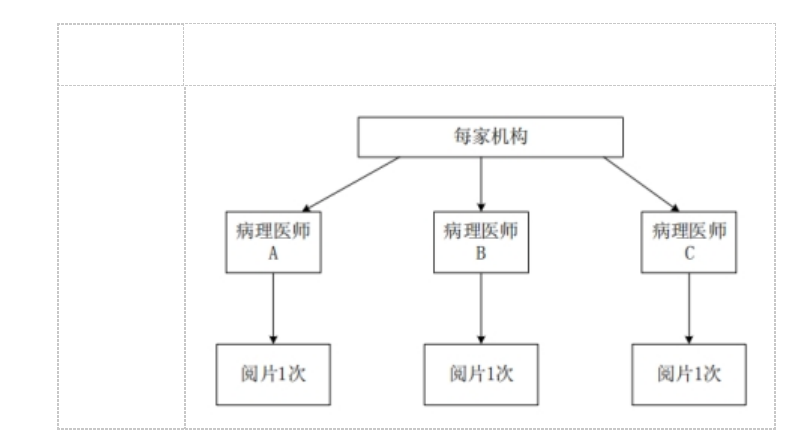
�����ڲ�ͬ����ҽʦ��Ƭһ�����о�Ӧ�������ٴ����������չ��ÿ���ٴ����������Ա��������ṩ���ٴ���������������Ⱦɫ�IJ�����Ƭ���б��о��������ٴ�����IJ�����ƬӦ�����ж����Ϊ���Ե���Ƭ���ж����Ϊ���Ե���Ƭ��
ÿ���ٴ��������ѡ��3����ͬ�����IJ���ҽʦ�����ٴ����飬�ٴ������������ÿ������ҽʦ�Ըû�����������в�����Ƭ�����ж������в���ҽʦ����Ƭ�����ɷ������ݼ��������ڲ�ͬ����ҽʦ��Ƭһ�������������ͼ3��
|
|
|
|
|
|
ͼ3.�����ڲ�ͬ����ҽʦ��Ƭһ�����������ʾ��ͼ
2.3��ͬ�����䲡��ҽʦ��Ƭһ����
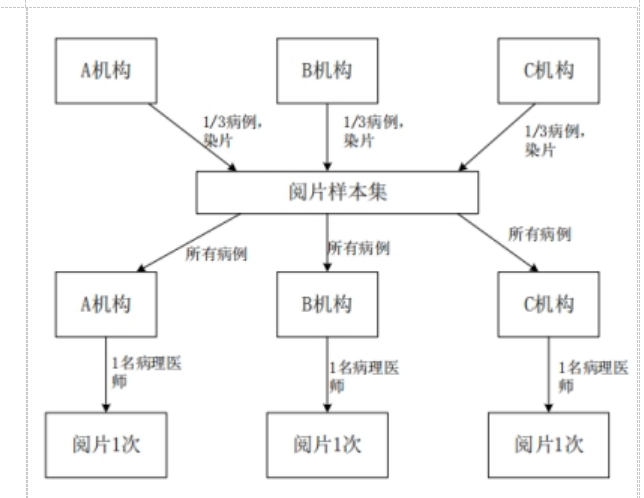
��ͬ�����䲡��ҽʦ��Ƭһ�����о�Ӧ�������ٴ����������չ���ٴ��������財����ƬΪ�����ٴ����������ͬ�ṩ����Ƭ��ɵ���Ƭ��������Ƭ�����������У�ÿ���ٴ���������ṩ��������Ƭ��������Ƭ������Ƭ��Ӧ��Ծ��⡣ÿ�Ų�����ƬӦΪ�����걨��Ʒ˵�������Ⱦɫ�IJ�����Ƭ��
���ٴ�������������ٴ������չ��չ��Ƭ������Ƭ������ÿ���ٴ�����������ȫ����Ƭ���ٽ���Ƭ��ת������һ�һ�������֤ÿ���ٴ���������о��߾��ܶ���Ƭ�������в����IJ�����Ƭ���ݲ�Ʒ˵����Ҫ����н���ж���
���ٴ���������յ���Ƭ����ѡ�����һ���������о��ߣ������걨��Ʒ˵�����Ҫ��Բ�����Ƭ���н���ж��������ٴ������������Ƭ�������в�����Ƭ�ж������ո��ṹ�������γ���Ƭһ�����о������ݼ������ں���ͳ�Ʒ�������ͬ�����䲡��ҽʦ��Ƭһ�������������ͼ4��
|
|
|
|
|
|
������������ѡ�������ռ�
������������Ƭһ�����о������������ӦΪ��ƷԤ��������Ⱥ��������۲�ƷPD-L1����Լ���Ԥ����;Ϊ���Լ�����Ը������̶ֹ�ʯ������FFPE����ʳ����״ϸ������ESCC����֯�е�PD-L1���ױ�������鲡��ӦΪ�ٴ���ȷ���Ϊʳ����״ϸ�����IJ�����
Ϊ�˸��ӿ�ѧ�����۲�Ʒ���ܣ��������о���Ӧ��������PD-L1����������ֵ�ٽ���Χ�ڲ���������ָ��������ҩ��ʹ�õ���ֵ�����ò��ֲ�����ռ����Ӧ���������Բ�����30%��
��������������Ա
�ٴ��������Ӧ��ù���ҩƷ�ල�����ֱ����Ͽɡ��ٴ��������Ӧ�����ϸ������������ϵ��ִ��ʵ�����ڲ��ճ��������ơ�
�ٴ�������Ӧ������������趨��ͬ�����IJ���ҽʦ�������飬����ҽʦ��������Ӧ������
���ģ�����������
�ٴ�����Ӧ���п�ѧ�����������㣬������ò������Ƶķ���������������������������̽����趨��������ˮƽ���������ɲ������¹�ʽ��
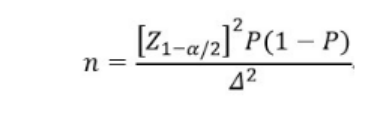
��ʽ��nΪ��������Z1-��/2Ϊ���Ŷȱ���̬�ֲ��ķ�λ����PΪ����ָ��Ԥ��ֵ����ΪP����������С��
���о��߲��������ĺ���������������ģ�ͣ��ڽ��к������͵�ǰ������ɲ��á�
1.��������
����������������Է�����Ӧ������85%�����Է�����Ӧ������85%������ˮƽȡ0.05���о���Ӧ�趨�����Ħ�ֵ���ݴ˹��㻷���������гɶԱȽϽ�������Բ����������Բ��������ڱ�֤��������������ǰ���£��ٴ������У���������������������������Ӧ����60�����������Բ�Ӧ����30�������Բ���������30�������������������������У��ṩ��������������������Ӧ�������⡣
2.��Ƭһ�����о�
2.1������ͬһ����ҽʦ��Ƭһ�����о�����ͬ����ҽʦ��Ƭһ�����о�
�ò����о�Ӧ��ÿ���ٴ��������Ϊ��λ���������������㡣���������������Ӧѡ����������Ҫ����ָ�꣬�������ο������һ�������ۻ�ɶԽ����һ�������ۡ����������Բ���ͬһ����ҽʦ�����Ƭ����ͳ�ƻ�ͬ����ҽʦ����ͳ�Ƶ�����һ���ʣ���ƽ������һ���ʣ�������һ���ʣ���ƽ������һ���ʣ��������Լ����Բ����Ĺ��㣬��������IJ���ӦΪ���㲡����1/3���������������Ӧ��ز������趨Ӧ��ѧ���������ò����о��ٴ����ܲ�Ӧ���ڻ������飬����ˮƽȡ0.05���о���Ӧ�趨�����Ħ�ֵ����Ԥ��ֵ����ʱ��Ӧ���Ǹ��ŵľ��ȡ�
2.2��ͬ��������Ƭһ�����о�
�ò����о�Ӧ�����һ����ṩ����Ƭ���������������㣬�����������������Ҫ����ָ���ѡ��ӦΪÿ���ٴ����������ο������������������Աȵ�����һ���ʡ�����һ���ʡ������������������ز������趨Ӧ��ѧ���������ò����о��ٴ����ܲ�Ӧ���ڻ������飬��Ӧ���ڻ�����ͬһ����ҽʦ��Ƭһ�����о�����ͬ����ҽʦ��Ƭһ�����о�������ˮƽȡ0.05���о���Ӧ�趨�����Ħ�ֵ����Ԥ��ֵ����ʱ��Ӧ���Ǹ��ŵľ��ȡ��ݴ˹��������ӦΪ���о���Ƭ������������Ƭ���������ٴ���������ṩ���������ṩ�����Լ�������Ƭ������Ӧ��Ծ��⡣
2.3�ڿ�ѧ������Ƶ�ǰ���£�������ͬһ����ҽʦ��Ƭһ�����о�����ͬ����ҽʦ��Ƭһ�����о�����ͬ��������Ƭһ�����о������о�����Ƭ���ظ�ʹ�á��ظ�ʹ����Ƭ������Ӧ����ͬһ����ҽʦ�ڶ�ʱ���ڶ�ͬһ����Ƭ����ж���
���壩����ָ�꼰ͳ�Ʒ���
�������鼰��Ƭһ�����о�ͳ�Ʒ���Ϊһ�������ۣ��ɲ����ĸ�������ķ�����������ο������һ�������ۼ��ɶԽ����һ�������ۡ��ο����һ��Ϊͬһ������ͬһ��Ⱦɫ��Ƭ�ڲ�ͬ��Ƭ���ж������У�����Ƶ�����Ľ������ο�����Աȹ�������Ҫ���ۿ����Լ���ο����������һ���ʣ�PPA��������һ���ʣ�NPA������һ���ʣ�OA�����ɶԽ����һ����������Ҫ�����о��������ɶԱȽϵĽ�����ɶ�һ��������ָ��Ϊƽ������һ���ʣ�APA����ƽ������һ���ʣ�ANA����ƽ����һ���ʣ�OA��������ͳ�Ʒ���Ӧ���ú����ķ���������������Ĺ��㡣
1����������
���������ͳ�Ʒ�����ҪΪ���ٴ�������������������ɶԱȽϣ��ɶԷ��������������飺����1�����2������2�����3������3�����1���Ե�һ�����1�����2�ɶԷ���Ϊ�����Ի���1�Ľ��Ϊ�����ĸ����ʽͳ�ƻ���2��������1�����һ���ʣ���������һ���ʼ�����һ���ʡ���������������������2�����3������3�����1��һ���ʡ����ս����������ܣ��������������ƽ������һ���ʡ�ƽ������һ���ʼ�ƽ����һ���ʡ�
2����Ƭһ�����о�
��Ƭһ�����о�ͳ�Ʒ���������ο������һ�������ۼ��ɶԽ����һ�������ۡ�
2.1 ������ͬһ����ҽʦ��Ƭһ�����о�
��ο������һ�������ۣ�ÿ�Ų�����Ƭ�IJο����Ϊ������ͬһλ����ҽʦ�����Ƭ����У�����Ƶ�����Ľ��������ҽʦ������Ƭ����ֱ���òο�������бȶԣ���������һ���ʡ�����һ���ʼ���һ���ʣ�������Ƭ�������Ϊ�û���ͬһ����ҽʦ��Ƭһ���ԣ����һ����������Ϊ���л���ͬһ����ҽʦ��Ƭһ�����о���
ͬһ����ҽʦ�ɶԽ����һ�������ۣ����۹��̰������飺��1����Ƭ���2����Ƭ����2����Ƭ���3����Ƭ����3����Ƭ���1����Ƭ���Ե�һ���1����Ƭ���2����Ƭ�ɶԷ���Ϊ�����Ե�1����Ƭ�Ľ��Ϊ�����ĸ����ʽͳ�Ƶ�2����Ƭ�Ľ�����1����Ƭ�����һ���ʣ���������һ���ʼ�����һ���ʡ�������������������2����Ƭ���3����Ƭ����3����Ƭ���1����Ƭ��һ���ʡ����ս����������ܣ�����������ͬһҽ����Ƭ��ƽ������һ���ʡ�ƽ������һ���ʼ�ƽ����һ���ʡ����һ����������Ϊ���л���ͬһ����ҽʦ��Ƭһ�����о���
2.2�����ڲ�ͬ����ҽʦ��Ƭһ�����о�
��ο������һ�������ۣ�ÿ�Ų�����Ƭ�IJο����Ϊ��������λ����ҽʦ��Ƭ����У�����Ƶ�����Ľ����������������ɲο�����2.1������ͬһ����ҽʦ��Ƭһ�����о���
�ɶԽ����һ�������ۣ����۹��̰������飺����ҽʦ1�벡��ҽʦ2������ҽʦ2�벡��ҽʦ3������ҽʦ3�벡��ҽʦ1��������������ɲο�����2.1������ͬһ����ҽʦ��Ƭһ�����о���
2.3�����䲡��ҽʦ��Ƭһ�����о�
��ο������һ�������ۣ�ÿ�Ų�����Ƭ�IJο����Ϊ��ͬ��������ҽʦ��Ƭ����У�����Ƶ�����Ľ����������������ɲο�����2.1������ͬһ����ҽʦ��Ƭһ�����о���
�ɶԽ����һ�������ۣ����۹��̰������飺����1��Ƭ��������2��Ƭ���������2��Ƭ��������3��Ƭ���������3��Ƭ��������1��Ƭ�����������������ɲο�����2.1������ͬһ����ҽʦ��Ƭһ�����о���
������ԭʼ����
1.�ύ�������ݻ��ܱ�������Ӧ���ٰ������Ա����䡢������Ͻ�����о����ж�����ȡ�
2.�ύ��ѡ����Ⱦɫ�����Բ�ɫͼƬ������Ⱦɫ�ص������֯��̬��Ⱦɫǿ�Ⱥͱ���Ⱦɫ�ص���м�Ҫ������
���ߣ�ƫ�п���
Ϊ�˿����ٴ������ƫ�У��������о�������Ӧ��Բ���/��ƬӦ������ä��ʹ�о�������������в�֪������������ϻ�������ؼ�����Ϣ���Ӷ���������ƫ�С�
�ڽ���ͬһ����ҽʦ��Ƭһ�����о������У�����ҽʦ�ڽ�����һ����ƬʱӦ����һ��ʱ��ļ�������ڣ������ڣ����˶�ʱ��һ�㲻�������ܡ�
ͬһ����ҽʦ��Ƭһ�����о������У�ÿ����Ƭʱ������Ϊ����һ�������IJ�ͬ�ĸ��Ų��������Ų���Ӧͬʱ�������Ժ����Բ����������Ų�������������У�ʹ�о���ͬʱ��Է������еIJ��������Ų������в����������Ų������������ͳ�Ʒ�����
���ˣ���������
�ٴ����鿪ʼǰ��������������ѵ����ȷ���о�����Ϥ������������鷽���IJ������������������ܵȣ�����ȿ�������������������̶�Ӧ������Ч�����������£�����ȱ�֤�������ݵ�ȷ�Լ����ظ��ԡ�
�ٴ�������Ӧ�ƶ��ϸ������ת�˷�����ȷ������ת�˹��̲����ٴ��������Ӱ�죬����ת�˹�����Ӧ�����������Ӽ�¼�����ٴ��������������Ӧ�ܹ���Դ��
�ٴ�����Ӧ�ϸ��ղ�Ʒ˵������в�����Ӧ�������Ҫ����б�Ҫ��HEȾɫ��Ӧ���ú������ա�
�ġ���ݵ�λ
����ҩƷ�ල������ҽ����е�����������ġ�
�塢�ο�����
1.����������Լ��ٴ����鼼��ָ��ԭ������ҩƷ�ල������ͨ��2021���76�ţ���2021��9��28�ա�
2.Xiao-Hua Zhou��Nancy A.Obuchowski��Donna K.McClish. ���ҽѧ�е�ͳ��ѧ�������ڶ��棩 [M].����:�ߵȽ��������磬2016��
3.����.ҽ����е�ٴ�����ͳ�Ʒ������ڶ��棩 [M].����:��ѧ�����磬2016��