

| ��ָ��ԭ��ּ��ָ��ע�������ˣ����¼�������ˣ����ǻ�Ѫ�쵰������ע���걨���ϵ�����д��ͬʱҲΪ����������������ע���걨�����ṩ�ο�����ָ��ԭ���Ƕ��ǻ�Ѫ�쵰�����ǵ�һ��Ҫ��������Ӧ���ݲ� |
��ָ��ԭ��ּ��ָ��ע�������ˣ����¼�������ˣ����ǻ�Ѫ�쵰������ע���걨���ϵ�����д��ͬʱҲΪ����������������ע���걨�����ṩ�ο���
��ָ��ԭ���Ƕ��ǻ�Ѫ�쵰�����ǵ�һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����á��������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ����
��ָ��ԭ���ǹ������˺�����������Աʹ�õ�ָ���ļ������漰ע������������������Ϊ����ǿ��ִ�У������ܹ����㷨��Ҫ�������������Ҳ���Բ��ã���Ӧ�ṩ��ϸ���о����Ϻ���֤���ϡ�Ӧ����ѭ��ط����ǰ����ʹ�ñ�ָ��ԭ��
��ָ��ԭ���������з��桢����ϵ����ǰ��֪ˮƽ���ƶ��ģ����ŷ��桢����ϵ�IJ������ƺͿ�ѧ�����IJ��Ϸ�չ����ָ��ԭ���������Ҳ����ʱ���е�����
һ�����÷�Χ
��ָ��ԭ�������ڲ������ӽ���ɫ��[������ѹҺ��ɫ�����ЧҺ��ɫ����HPLC��������ѹҺ��ɫ����LPLC����]������ѪҺ��HbA1cŨ�Ƚ��м��ķ����ǡ��Ի�������������HbA1c�����Dz�Ʒ���ɲ��ձ�ָ��ԭ�������������HbA1c����ֻ�������÷�Χһ���ֵ��ٴ�����������Ӧ�����ձ�ָ��ԭ����HbA1c�������ֵ����ע���걨���ϡ�
����ע�����Ҫ��
��һ�������Ϣ
1.��Ʒ����
��Ʒ����Ӧ���ϡ�ҽ����еͨ����������������ҵ����ͨ�����Ƶ�Ҫ��Ʒ����ͨ����һ�����ĴʺͲ�������������������ɣ��������ֲ�Ʒ�����ṹ���������������Լ��Զ����̶�Ϊ���������磺�ǻ�Ѫ�쵰�����ǡ�ȫ�Զ��ǻ�Ѫ�쵰�����ǵȡ�
2.�������
�ǻ�Ѫ�쵰�����ǣ����ա�ҽ����е����Ŀ¼������Ʒ�Ĺ������Ϊ���࣬��Ʒ�������Ϊ22-10-04��
3.ע�ᵥԪ����
ע�ᵥԪ����ԭ�����Բ�Ʒ�ļ���ԭ�����ṹ��ɡ�����ָ������÷�ΧΪ�������ݡ��ǻ�Ѫ�쵰������ע�ᵥԪ���ֽ����ص��ע���·��档
3.1����ԭ��
��ͬ����ԭ���IJ�ƷӦ����Ϊ��ͬ��ע�ᵥԪ����HPLC����LPLC����
3.2�Զ����̶�
��Ʒ�Զ����̶Ȳ�ͬ��Ӧ����Ϊ��ͬ��ע�ᵥԪ������Զ���ȫ�Զ���
��������������
1.�ṹ���
�����˿ɸ��ݲ�Ʒ�ľ�������������Ʒ�ṹ��ɣ�������������Լ����ʹ�õĸ�����������ͼƬ������ֵ���ʽ����˵����
�ǻ�Ѫ�쵰�����ǣ����¼�Ʒ����ǣ�ͨ���ɽ���ģ�顢����ģ�飨ɫ�����������ģ�顢�����������ɡ�
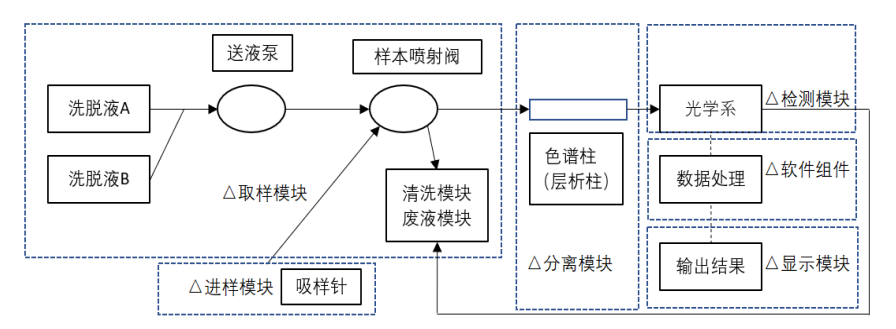
ͼ1 ��Ʒ�ṹ���ʾ��ͼ������
2.��ɲ���������
2.1 ����ģ�飺ͨ���������롢��ϴģ�顢��Һģ�����ɡ�
2.2����ģ�飺��Ҫ��ɫ����������������ɡ���Ԥ��������Ʒͨ������ģ�飬������Ʒ��ϴ��Һ�Լ�ɫ����������������������֮���ͳ̶ȣ�������HbA1c���ڸ��ɷֵ�ϴ��˳��
ע��Ϊ��ȷ������ʱ���¶ȣ�Ҳ���������м��²�����
2.3���ģ�飺��Ҫ������Դ����LED����������ܣ������������Ԫ����ȡ�ͨ������ɫ�����������������������и���ֵ������ֵ��������ת�����źţ������γ�ɫ�ס�
2.4������������ü�ɫ�ĸ������������ɸ��ɷֵ�Ũ�ȣ��䱨��������HbA1c�ⶨֵ�Լ�ɫ��ͼ����һ�������õ�λ���ֳ�NGSP��λ����%�����ǻ�Ѫ�쵰�ײⶨ�����ͬʱ������ʵ�λ���ֳ�IFCC����mmol/mol�Ľ����
2.5Ӧ��ϸд��ͨ���о�ȷ���Ĺؼ������Ϣ�������������Ʋ��������ݡ�
3.��Ʒ����ԭ��
���ӽ���ɫ��[���ѹҺ��ɫ�����ЧҺ��ɫ����HPLC��������ѹҺ��ɫ����LPLC����]��
ͨ������HbA1c������Ѫ�쵰����֣�HbA1a��HbA1b��HbF��HbA0��������ɲ�ͬ����һ���Ļ���Һ����ǿ���¿���ɫ����������������֬����ż�������ӻ��Ų�ͬ�̶ȵĽ�ϣ��Ӷ���HbA1c������������ֿ������룬����ɫ��������������ϴ�Ѹ��ɷֵ�����ȣ�����ÿ����ֵ�µ��������HbA1cռ��Hb�ı������Ӷ���÷����ͺ�����
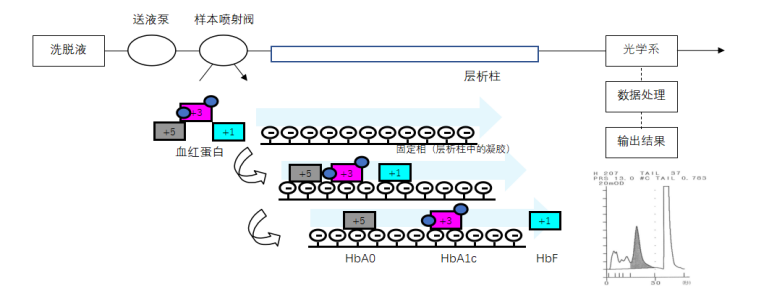
ͼ2 ���ӽ���ɫ������ԭ��ͼ������
4.���÷�Χ�ͽ���֤
��Ʒ���÷�ΧӦ���걨��Ʒ�IJ�Ʒ���ܱ���һ�¡�
�����ǵ����÷�Χ�����걨��Ʒ�����ԶԲ�Ʒ�Ĺ���ԭ���Լ�Ԥ��ʹ�û���������ȷ��һ�������Ϊ���ò�Ʒ�������ӽ���ɫ���������ļ���Լ���ͬʹ�ã����ٴ������ڼ������ȫѪ�����е��ǻ�Ѫ�쵰������
���걨��Ʒ���÷�Χ�а���Ѫ�쵰��A2��HbA2����̥��Ѫ�쵰�ף�HbF���Լ��쳣Ѫ�쵰�ף�HbC��HbD��HbE��HbS�ȣ��ļ�⣬��������Ӧ�����ձ�ָ��ԭ������Ҫ���ύ�о����ϡ�
5.�ο���ͬ���Ʒ��ǰ����Ʒ�����
Ӧ���ṩͬ���Ʒ�������������У���ǰ����Ʒ����Ϣ����������ע���Ʒ���з�������Ŀ�ġ�����ͬ���Ʒ��Ӧ��˵��ѡ������Ϊ�з��ο���ԭ��ͬʱ�б�˵������ע���Ʒ��ο���Ʒ��ͬ���Ʒ��ǰ����Ʒ���ڹ���ԭ�����ṹ��ɡ�����ָ���Լ�Ԥ����;�ȷ������ͬ����Ҫʱ�ṩͼʾ��
���������ٴ�����
1.��Ʒ���շ���
��Ʒ��Ҫ�ķ��հ�������Σ��������Σ������ʹ���йص�Σ��������ʧЧ���ϻ��йص�Σ���ȣ�Ӧ����YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á���Ҫ���ÿ�ֿ����漰��Σ��ʶ���������γɷ��շ����������档��ʼ�¼��ͻ���ʾ��������1��
2.ҽ����е��ȫ�����ܻ���ԭ��
�����˿ɸ��ݲ�Ʒ�����ж�ҽ����е��ȫ���ܻ���ԭ���и�����������ԣ�֤�������Բ��õķ�����Ϊ�������ṩ��֤�ݵ��ļ��������˸���ʵ�����������д��
֤����Ʒ�����Ե��ļ��������շ������ϡ��о����ϡ��ٴ��������ϡ����鱨�漰������ϵ�����ļ��ȡ��������걨�����е�֤���ļ���Ӧ��˵�������λ�á�����������ļ�ʱӦ���嵽�ύ�ĵ����ļ����������о������еIJ��Ա��棨������XXX������������ͬһ�ļ���ʱӦ���嵽����Ż�����ţ�����շ������Ϸ�������������X.X��δ�������걨�����е�֤���ļ���Ӧ��ע���ļ����Ƽ����ļ���ű��顣
3.�����
3.1��Ʒ�����о�����
3.1.1������ָ���о����ϣ�
�걨��Ʒ�����ģ�����ܵ��о����ϣ�Ӧ���������������й��걨��Ʒ�ṹ��ɺ���Ҫ���ģ���������ṩ��ϸ���о����ϣ�ͨ��Ӧ��������ģ�顢����ģ�飨ɫ�����������ģ�顢��������Ĺ�����ָ�����ģ������ҪԪ����������ָ����о����ϡ�
3.1.2��ȫ��ָ�����֤����������ȫָ��͵�ż���ָ�������ࡣ������ȫָ��Ӧ������GB 4793.1�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��1���֣�ͨ��Ҫ������GB 4793.6�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��6���֣�ʵ�����ò��ϼ����豸������Ҫ������������ģ���������ȡ����������������ã���GB 4793.9�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��9���֣�ʵ�����÷���������Ŀ�ĵ��Զ��Ͱ��Զ��豸������Ҫ������YY 0648�����������ƺ��������õ����豸�İ�ȫҪ�� ��2-101���֣�������ϣ�IVD��ҽ���豸��ר��Ҫ�������������õĹ��ұ�����ҵ���е�����ָ�ꣻ��ż���ָ��Ӧ������GB/T 18268.1���������ƺ�ʵ�����õĵ��豸��ż�����Ҫ���һ���֣�ͨ��Ҫ������GB/T 18268.26�����������ƺ�ʵ�����õĵ��豸��ż�����Ҫ�� ��26���֣�����Ҫ�� ������ϣ�IVD��ҽ���豸�����������õĹ��ұ�����ҵ���е�����ָ�ꡣ�߱������������˿ɶ�������Ŀ�����о������ύ��ϸ����֤���ϣ����߱������������˿�ͨ�����鱨���������Ŀ������֤���Լ��鱨����Ϊ�ò��ֵ���֤���ϡ�
3.2��Ʒ��Ч�ںͰ�װ�о�
Ӧ���ṩ��Ʒ��Ч�ڵ���֤���棬������Ӧ���걨��Ʒ�а������ġ������趨�ڸ������߾��й̶�ʹ����������ҪԪ���������������ϸ����������ȷ����Ʒʹ��������ʧЧ�ڵľ������ɣ�������Ʒʹ�������߲�ƷʧЧ�ڡ�����μ�����Դҽ����еʹ������ע�Ἴ�����ָ��ԭ�����Ҫ��
��װ����װ�����ԣ������Ƶ���Ч�����Լ����䴢�������£����ְ�װ�����Ե����ݡ�
3.3�����о�
3.3.1Ӧ���ҽ����е�����о����Ͽ���ύ��Ӧ���о����ϣ������İ�ȫ�Լ�������������Ԥ����;��ʹ�ó��������Ĺ����ۺ��ж���
ͼ3 ҽ����е�����о����Ͽ��
3.3.2�����汾����������ȷ���������汾ȫ���ֶε�λ������Χ�����壬������ģ�飨��ҽ���м�����������а汾���������ṩ��汾����������ȷ�������汾��������Ĺ�ϵ������������ģ��İ汾�����������������������ϵ����һ�¡�
3.3.3�й������о����ϵ���ϸ���ݣ�Ӧ���ա�ҽ����е����ע�����ָ��ԭ��2022�����棩��Ҫ����б�д��
3.4���簲ȫҪ��
��Ʒ�漰ҽ����е���簲ȫ��ָ�߱��������ݽ�����Զ�̷�������ơ��û��������ֹ��ܵ���һ�ּ����Ϲ��ܣ������������������ֳ����������У�����������ߡ��������磬�������ݽ��������������硢�洢ý��ĵ���˫�����ݴ��䣬Զ�̷�������ư������������ʵʱ����ʵʱ�ķ�������ƣ��û�����ҽ����Ա�����ߡ�ά����Ա�ȣ����ʰ������������û����桢���ӽӿڵ��˻�������ʽ��
Ӧ���ҽ����е���簲ȫ�о����Ͽ���ṩ��Ӧ���о����ϡ�
ͼ4 ҽ����е���簲ȫ�о����Ͽ��
�й����簲ȫ�о����ϵ���ϸ���ݣ����鰴�ա�ҽ����е���簲ȫע�����ָ��ԭ��2022�����棩����Ҫ����б�д��
3.5������ȫ��Ч�Ե��о�����
3.5.1������Ӧ�ύ����ϵͳ���������������о����ϣ��������ԡ�ȷ�ȡ����ܶȡ������Ե���Ŀ�������������о�Ӧ������HbA2��HbF�Լ��쳣Ѫ�쵰�ף�HbC��HbD��HbE��HbS�ȣ��Բⶨֵ��Ӱ�졣
3.5.2����ע���Ʒ�����÷�Χ��������Ƴ��˿ɱ����ǻ�Ѫ�쵰�ף�HbA1c���ⶨ����⣬���ɱ���HbA2��HbF���Լ��쳣Ѫ�쵰�ף�HbC��HbD��HbE��HbS�ȣ��IJⶨ�����������Ӧ�����Ǽ����Ŀ����ȷ�ȡ����ܶȵȵ��о����������HbA2��HbF����Ӧ�����Ǽ����Ŀ�������Ե��о��������о���Ŀ��Ӧ�ڲ�Ʒ����Ҫ������ȷ������õ�Ҫ��
4.��Ʒ����Ҫ��
��Ʒ����Ҫ�������Dz�Ʒ��Ҫ��������ָ�����������Ҫ�Ļ���֮һ��
4.1����ָ��
4.1.1�ɲο�YY/T 1246���ǻ�Ѫ�쵰�����ǡ����ã�������Ӧ��ϲ�Ʒ�������ܡ�����ԭ���ص㡢����ˮƽ���Կ������õı�����Ʒ����Ҫ��������ָ����Ŀ�ľ���Ҫ��Ӧ�������о����ϱ���һ�£�������ȷ�����о��������ݡ�
4.1.2�����ǻ�������ָ��
�����ǻ�������Ҫ��Ӧ���Ʒ�����ļ����ص�һ�£���Ӧ��ʹ��˵��������ȷ˵����ع��ܡ�
4.1.3ɫ��������������
����ע���Ʒ������ɫ����������������������ָ�����ȷ�����Լ����������Ҫ���⣬�����˻�Ӧ������ɫ�������������������Ҫ������Ч�ܡ���ѹ�����µȡ�
4.1.4��ȫҪ��
����GB 4793.1�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��1���֣�ͨ��Ҫ������GB 4793.9�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��9���֣�ʵ�����÷���������Ŀ�ĵ��Զ��Ͱ��Զ��豸������Ҫ������YY 0648�����������ƺ��������õ����豸�İ�ȫҪ�� ��2-101���֣�������ϣ�IVD��ҽ���豸��ר��Ҫ���������������Ҫ��
ע��������ģ���������²��������ȡ��������ܣ�����Ӧ����GB 4793.6�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��6���֣�ʵ�����ò��ϼ����豸������Ҫ������Ҫ��
4.1.5��ż���Ҫ��
����GB/T 18268.1���������ƺ�ʵ�����õĵ��豸��ż�����Ҫ���һ���֣�ͨ��Ҫ������GB/T 18268.26�����������ƺ�ʵ�����õĵ��豸��ż�����Ҫ�� ��26���֣�����Ҫ�� ������ϣ�IVD��ҽ���豸�������������Ҫ��
ע����ż����������е���Ʒ����ģʽӦ��������ع���ģʽ��
4.1.6��������Ҫ��
���ա�ҽ����е����ע�����ָ��ԭ��2022�����棩�������Ҫ��
4.1.7���簲ȫ
���ա�ҽ����е���簲ȫע�����ָ��ԭ��2022�����棩�������Ҫ��
5.���鱨��
ͬһע�ᵥԪ��������IJ�ƷӦ���ܹ�������ע�ᵥԪ��������Ʒ�İ�ȫ�Ժ���Ч�ԡ���ͬһע�ᵥԪ�ڴ�����Ʒ��ѡȡӦ���Dz�Ʒ���÷�Χ������ָ�ꡢ��ȫָ�ꡢ�ṹ��ɵȣ�����ԭ�����£�
5.1����Ӧ���������ԭ��ȷ�������ܸ��ǵIJ�����Ӧ�ֱ���м�⡣
5.2���漰�ؼ�Ԫ������һ�£���������Ӧ�����ǰ�ȫ�ؼ�Ԫ������һ�¶Ե�����ȫ�����Լ���ż���Ҫ���Ӱ�졣
5.3��û�г���֤���ܹ�֤��ͬһע�ᵥԪ�ڲ�ͬ�ͺŹ���Ʒ֮���ż����Կ��Ը���ʱ��Ӧѡȡÿһ�ͺŹ���Ʒ���е�ż�����Ŀ��⡣
���ڴ�����Ʒ��ѡ��������Ӧ���ṩ�����������֤����
���ģ��ٴ���������
���ݡ�����ҩ��ֹ��ڷ��������ٴ�����ҽ����еĿ¼��ͨ�桷�����¼�ơ�Ŀ¼����������������ע���Ʒ�����ǻ�Ѫ�쵰�ף�HbA1c��Ũ�ȼ�⣬����������ύ�ٴ��������ϡ�
������ע���Ʒ���÷�Χ�а���Ѫ�쵰��A2��HbA2����̥��Ѫ�쵰�ף�HbF���Լ��쳣Ѫ�쵰�ף�HbC��HbD��HbE��HbS�ȣ��ļ�⣬����֤���걨��Ʒ�롶Ŀ¼����Ʒ���е�ͬ�ԣ���Ӧ���ա�ҽ����е�ٴ����ۼ���ָ��ԭ������ҩƷ�ල������ͨ��2021���73�ţ���Ҫ��չ��Ӧ������
���壩��Ʒ˵����ͱ�ǩ����
��Ʒ˵���顢��ǩ�Ͱ�װ��ʶ�ı�дӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨����YY/T 0466.1��ҽ����е ����ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���֣�ͨ��Ҫ��GB/T 191����װ����ͼʾ��־����GB 4793.1�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��1���֣�ͨ��Ҫ��GB 4793.6�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��6���֣�ʵ�����ò��ϼ����豸������Ҫ�������ģ��������ȡ��������ܣ���GB 4793.9�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��9���֣�ʵ�����÷�������������YY 0648�����������ƺ��������õ����豸�İ�ȫҪ�� ��2-101���֣�������ϣ�IVD��ҽ���豸��ר��Ҫ��GB/T 29791.3���������ҽ����е �������ṩ����Ϣ����ʾ����3���֣�רҵ�������������������ر����Լ���ҽ����е����ע�����ָ��ԭ��2022�����棩������ҽ����е���簲ȫע�����ָ��ԭ��2022�����棩���ж�˵��������Ҫ��
˵���顢��ǩ������Ӧ����ʵ����������ѧ�������Ʒ������һ�£��������ݱ���ʹ�����ģ����Ը����������֡����ĵ�ʹ��Ӧ�����Ϲ���ͨ�õ��������ֹ淶��ע��֪ʶ��Ȩ��˵���顢��ǩ����װ��ʶ�е����֡����š�ͼ�Ρ��������֡�ͼƬ��Ӧ�һ�£���������ر��淶Ҫ��
1.˵����
ÿ̨�豸��Ӧ����˵���顣
˵�����������ٰ�����ҽ����е˵����ͱ�ǩ�����涨���е�ʮ�����йع涨Ҫ��ͬʱ��������˵���黹Ӧ�����������ݣ�
��1��ע�����������Ҫ��ʾ������ʾ�����ݣ��������쳣����Ĵ����������Լ��������浥�������쳣ɫ�ף��Ľ��͡�
��2����ע�ᵥԪ�а���ɫ����������������Ӧ��ȷɫ���������������ɼ������������Ϣ���ﵽ������Ӧ������Ӧ��ʩ�����ɳ���ʹ�á�
��3�������ǵ�ά���ͱ��������Լ������ų�������������������������·©Һ���������ݣ�����Ѫ�쵰��Ũ�ȹ��ߣ�����е���ϵȴ�����������
��4��GB/T 18268.26�����������ƺ�ʵ�����õĵ��豸��ż�����Ҫ�� ��26���֣�����Ҫ�� ������ϣ�IVD��ҽ���豸�� ����9 Ҫ�����Ҫ��˵���������������ݡ�
2.��ǩ����Ǻ��ṩ��Ϣ�ķ���
��1�����ձ�GB/T 191����װ����ͼʾ��־����YY/T 0466.1��ҽ����е ����ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���֣�ͨ��Ҫ��������飬˵������Ӧ����ر�־��ͼʾ˵����
��2����Ʒ�ı�ǩ���ⲿ������漰��ȫʹ�õIJ���Ӧ����GB 4793.1�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��1���֣�ͨ��Ҫ������GB 4793.6�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��6���֣�ʵ�����ò��ϼ����豸������Ҫ�������ģ��������ȡ��������ܣ���GB 4793.9�����������ƺ�ʵ�����õ����豸�İ�ȫҪ�� ��9���֣�ʵ�����÷���������Ŀ�ĵ��Զ��Ͱ��Զ��豸������Ҫ������YY 0648�����������ƺ��������õ����豸�İ�ȫҪ�� ��2-101���֣�������ϣ�IVD��ҽ���豸��ר��Ҫ������Ҫ��
���������
[1]ҽ����е�ල��������[Z].
[2]ҽ����е˵����ͱ�ǩ�����涨[Z].
[3]ҽ����еͨ��������������[Z].
[4]ҽ����еע���뱸�������취[Z].
[5]�ֹܾ��ڷ���ҽ����е����Ŀ¼�Ĺ���[Z].
[6]�����ٴ�����ҽ����еĿ¼��2021�꣩[Z].
[7]���ڹ���ҽ����еע���걨����Ҫ�����֤���ļ���ʽ�Ĺ���[Z].
[8] ���ڷ���ҽ����е��Ʒ����Ҫ���дָ��ԭ���ͨ��[Z].
[9]ҽ����е���簲ȫע�����ָ��ԭ��2022�����棩[Z].
[10]ҽ����е����ע�����ָ��ԭ��2022�����棩[Z].
[11]YY/T 1246���ǻ�Ѫ�쵰������[S].
[12]WS/T 461���ǻ�Ѫ�쵰���[S].
[13]���裬����ţ���ǻ�Ѫ�쵰���ٴ���ⷽ����չ�о�[J].���ʼ���ҽѧ��־��2012,33(8):954-956.
����1
�ǻ�Ѫ�쵰�����Ƿ��շ�������Ҫ��
��Ʒ���չ������������� YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á������Ҫ������ͨ���ο�ISO 14971�����Բ�Ʒ��ص�Σ�գ�Դ������Ԥ�ƺ������������ط��յĿ��ƺͷ��տ��Ƶ���Ч�ԡ����������ṩע���Ʒ�ķ��չ�������ʱ��Ӧ��Ҫ˵���������ݣ�
��1���ڲ�Ʒ�Ŀ����Σ���DZ��Σ���Ϳ��ܷ����ķ��ս���Ԥ�ƺ���������ȷʵʩ���ͷ��յļ������������ʩ��
��2����Ʒ�����ܼ���У�������֤�˸ô�ʩ����Ч�ԣ���������ͨ�ú���Ӧר�ñ���Ҫ��
��3����������ʣ����ա�
��4��ȫ���ﵽ�ɽ��ܵ�ˮƽ��
��5���Բ�Ʒ��ȫ�Եij�ŵ��
1.���չ����������������Ӧ�����������ݣ�
��1����Ʒ�ķ��չ�����֯��
��2����Ʒ����ɺ����÷�Χ��
��3�����ձ������������ݡ�
��4����Ʒ��ȫ������������ж���
2.�ɲο��ĸ�¼
��1�����Ʒ�йصİ�ȫ�������жϿɲο�YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á��ĸ�¼C��H��
��2��Σ�գ�Դ������Ԥ�����¼����к�Σ������ɲο�YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á���¼E��H��I��
��3�����տ��Ƶķ�����ʵʩ���ۺ�ʣ����յĿɽ��������ۼ������������������ط����ɲο�YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á���¼F��G��H��J��
��4�����տɽ������ͷ��յĴ�ʩ����ȡ��ʩ����յĿɽ��̶ܳȣ��Ƿ����µķ��ղ�����
��5���ǻ�Ѫ�쵰�����dz����ij�ʼ�¼��ͻ���ʾ��������1��
��1 ��ʼ�¼��ͻ���ʾ��
|
ͨ����� |
��ʼ�¼��ͻ���ʾ�� |
|
�������� Ҫ�� |
��Ʋ����IJ�ǡ���淶���ɴ����������֡���ǵ�����粿�ָ���/�������ȱ�ݣ����µ��Σ�շ����ϵͣ����ܶ�ʹ������ɵ��Σ�����豸��ͷ�IJ������ڲ����������յ�ɹ��ߣ�����̨֧�ż�ǿ�Ȳ��㣬�豸�桢�ǡ��ߴֲڣ����ܶ�ʹ������ɻ�е���ˣ��ܳ������������㣬��ɵ��Σ�����˶���������ʧЧ����ɻ�еΣ������ż����Բ�����Ҫ�����豸��������������������������豸�������������ܵ�©Һ���������豸��ˮ�����µ��Σ���� ���ܲ�����ǡ���淶��ȷ�ԡ��ظ��ԵȲ�����Ҫ�� ��еģ��ʧЧ������������������ȷ�����ʹ���λ��ȷ�����¼�����쳣�� ���Һ������ص����ﰲȫ�����⡣ �����е�Ҫ��ǡ���淶��˵����δ���ǻ�Ѫ�쵰�����ǵ�ʹ�÷������豸��ά������������˵���� Ԫ�������������������ʧЧ����ʾ���ϡ���ӡ���Ϻ�����������豸��������������ȫ���ܳ��������� |
|
������� |
���Ƴ�������������δ����֤�����²�Ʒ�����ܲ���������Ҫ�� ���������йؼ�������Ƶ�δ���м�⣬���²������������ϸ� �����Ŀ��Ʋ���֣��������Э������ѡ�����������Э��δ������Ч��������ȡ� |
|
��������� |
���ʵ��İ�װ����Ʒ�������������豸����������� ��ǡ���Ļ��������ȣ��ڳ����豸�涨�����滷�����¶ȡ�ʪ�ȡ�����ѹ����Ҫ���£������豸�������������� |
|
�������� |
����ѧ�ģ����ȡ�ѹ����ʱ�䣩�����䡢���ȵĻ������ܵ����豸��������������δ��ʹ�û��������������ϸ���֤������ʹ�������IJ���Ӧ�����¼������ȷ�� ��ų�����Ե�Ÿ��ŵ����жȣ�������Ÿ���������ض��������豸������������A���豸��B���豸�Ļ�����ʹ�û�Թ�����������Ӱ�죬���Ź��������������õ��豸���������С� ���ʵ���������Ӧ���豸�Ĺ����ѹ���ȶ��������豸�����������������������ȷ�� |
|
��ࡢ��������� |
δ�Գ�ϴ�������̽���ȷ�ϻ�ȷ�ϳ��淶�� ʹ����δ��Ҫ����г�ϴ�����ȡ� |
|
���úͷ��� |
û�ṩ��Ϣ���ṩ��Ϣ����֣�δ�ṩ��Ʒʹ�ú��û���ʾ����ֵȡ� |
|
������ |
���ȱ��������ʹ�ô���ȡ� ����������ʹ��˵���� ��ȱ����ϸ��ʹ�÷�����ȱ�ٱ�Ҫ�ļ���������ȱ�ٱ�Ҫ�ľ���˵����ȱ����������滷�����������ƣ��豸�ڹ���״̬�����ѹ�����ء��Ͽ������ӵ��ߡ��豸��Ԫ�������ֹ��ϣ������пɲ���Σ�վ�ʾ���㣻ʹ��ǰδ����豸����״̬������˵�����ڸ��ӣ�������δ˵�������ȷά���������豸/������ ��ϴ����������ȷ�������� ���û��������δ��ʾ��λ�� ����������Σ���ԣ���Ա�������⡣ �������������Ӧ����ʾ��Ϣ��ʵ��״̬����Ӧ�� ��ȱ��������/δ����ѵ����Աʹ�ã�ʹ����/������δ����ѵ����ѵ���㣬������ȷʹ�ú�ά���������豸�� |
|
ʧЧģʽ |
�����ϻ���ĥ����ظ�ʹ�ö����¹����˻�/ƣ��ʧЧ�ȡ� |