

| ��ָ��ԭ��ּ��ָ��ע�������˶Գ�����������Ѫ�������Dz�Ʒע���걨���ϵ�����д��ͬʱҲΪ����������������ע���걨�����ṩ�ο�����ָ��ԭ���ǶԳ�����������Ѫ�������ǵ�һ��Ҫ��������Ӧ���ݲ�Ʒ |
��ָ��ԭ��ּ��ָ��ע�������˶Գ�����������Ѫ�������Dz�Ʒע���걨���ϵ�����д��ͬʱҲΪ����������������ע���걨�����ṩ�ο���
��ָ��ԭ���ǶԳ�����������Ѫ�������ǵ�һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ����������Ƿ����ã��������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ����
��ָ��ԭ���ǹ�ע�������˺�����������Աʹ�õ�ָ���ļ������漰ע������������������Ϊ����ǿ��ִ�У������ܹ����㷨��Ҫ�������������Ҳ���Բ��ã���Ӧ�ṩ��ϸ���о����Ϻ���֤���ϡ�Ӧ����ѭ��ط����ǰ����ʹ�ñ�ָ��ԭ��
��ָ��ԭ���������з��桢����ϵ����ǰ��֪ˮƽ���ƶ��ģ����ŷ��桢����ϵ�IJ������ƺͿ�ѧ�����IJ��Ϸ�չ����ָ��ԭ���������Ҳ����ʱ���е�����
һ�����÷�Χ
��ָ��ԭ�������ڶԾ�������������Ѫ�ܵ�Ѫ�������ij�����������Ѫ�������ǡ����ݡ�ҽ����е����Ŀ¼���е�ҽ�����ͼ��е���������ΪII�࣬��Ʒ�������Ϊ07-07-01��ҽ�����ͼ��е-�����������������������豸-����������Ѫ�������豸����
���Ʒ�����÷�Χ����������Ѫ�ܵ�Ѫ���������ɲ��ձ�ָ��ԭ���������ִ�С�������������Ѫ��������ͨ��ʹ�õ��ǵ�Ԫ̽ͷ��������Ƭ̽ͷ��PW��ʽ����˫��Ƭ̽ͷ��CW��ʽ����������������̽ͷ�IJ�Ʒ�������ڱ�ָ��ԭ��
ע�������ʽ�豸�У����Ӳ���Ӧ������Ӧ��ר�ñ�����ָ��ԭ��δ�漰���Ҫ��
����ע�����Ҫ��
��һ�������Ϣ
1.��Ʒ����Ҫ��
��Ʒ������Ӧ���ϡ�ҽ����еͨ��������������Ҫ���á�ҽ����е����Ŀ¼������ұ�����ҵ���е�ͨ�����ƣ����磺������������Ѫ�������ǡ�����������Ѫ������ǵȡ�
2.ע�ᵥԪ���ֵ�ԭ���ʵ��
������������Ѫ�������ǵ�ע�ᵥԪԭ�����Խṹ��ɡ�����ָ������÷�Χ����Ϊ�������ݡ�
2.1��Ʒ�ṹ����нϴ����ij�����������Ѫ��������Ӧ����Ϊ��ͬ��ע�ᵥԪ��������������ͼ1��ͼ2��ʾ�ij�����������Ѫ��������Ӧ����Ϊ��ͬ��ע�ᵥԪ����ͬ����������͵ij�����������Ѫ�������������ṹ����ϴ�ҲӦ��Ϊ��ͬע�ᵥԪ����ע�ᡣ�����������ͷֱ�Ϊ���ࣨ����ڲ���Դ�������ࣨ����ڲ���Դ���ͽ�Ϊ�ڲ���Դ����ij�����������Ѫ�������ǣ�Ӧ����Ϊ��ͬ��ע�ᵥԪ��
2.2��Ҫ����ָ�겻�ܸ��ǡ��нϴ����ģ�Ӧ���ֲ�ͬ��ע�ᵥԪ��
2.3��ijһ�ͺŵ����÷�Χ���Ը��������ͺŵ����÷�Χ�⣬��ͬ���÷�Χ�ij�����������Ѫ��������Ӧ����Ϊ��ͬ��ע�ᵥԪ��
��һ����������
1.��Ʒ�Ľṹ�����
������������Ѫ��������ͨ��������������̽ͷ�����������������ã���ȷ�����汾����ͨѶ���£������ã�����Դ�ߣ������ã����ͷ�ף������ã���̨���������ã���ң�����������ã�����̤���أ������ã���ɡ�
2.��ɵ�Ԫ�ṹ/��������
��1����������Ҫ�����źŲɼ�ģ����ͼ����ģ���ȣ����źŲɼ�ģ�������Դת�����������շ����źŷŴ��ƽ��������ѡͨ��AD�����٣����������ݴ�������ݣ���Ҫʵ���˳������ķ�������ա�ģ��ת�������ݴ洢�봫��ȹ��ܣ�ģ�����ʽ��Ʒͼ����ģ���ɼ��������ִ�С�
��2������̽ͷ��ͨ��Ϊ��Ԫ̽ͷ���ʽ̽ͷ�����岨��PW��̽ͷʹ�õ��ǵ���Ƭ̽ͷ����������CW��̽ͷʹ�õ���˫��Ƭ̽ͷ����Ҫ�ǽ��г������ķ�������գ������յ��Ļز��źŴ�����������
��3��ͨѶ���£������ã��������źŲɼ�ģ��͵���ϵͳ֮������ݴ��䣬ͨ��ΪUSB�ӿڣ�Ҳ��ʹ������ͨ�Žӿ���ʽ���д��䡣
��4����Դ�ߣ������ã�����������Դ���缰�����������ӣ�Ҳ�ɺ���Դ��������
��5���ͷ�ף������ã������ڹ̶�̽ͷ����������ͷ����������ⱻ��Ѫ�ܵ�Ѫ��������һ���Ϊ�ֶ����Զ����֡�
3.��Ʒ���������ʵ��
����Ʒ��̬һ���Ϊ����Яһ��ʽ��ģ�����ʽ��
��ͨ������Ϊ����ͨ����˫ͨ������ͨ������
��һ��ͨ������ͬʱ��õ�������������Է�Ϊ������ȡ�����ȣ�

ͼ1������������Ѫ�������ǣ�ģ�����ʽ��
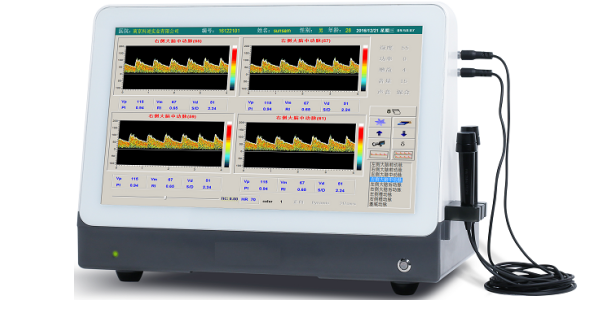
ͼ2������������Ѫ�������ǣ���Яһ��ʽ��
4.��Ʒ����ԭ��
������������Ѫ��������ͨ�������ּ�ⷽʽ�����岨����������ʽ�����岨�����ڼ����Ѫ�ܣ��о���ѡͨ�����������������ڼ�⾱��Ѫ�ܺ�����Ѫ�ܣ�����ѡͨ������
���������廹��������������ԭ����������ͨ��̽ͷ�����巢�䳬����������ѪҺ���˶���Ѫ��ϸ���Գ������ķ������ã������ز��źš���ͬ�˶��ٶȵ�Ѫ��ϸ���������ͬƵ�ʵĶ�����Ƶ�ơ�����ͨ�����ܻز��źţ������ź�Ƶ�Ƽ��ɼ����Ѫ��ϸ�����˶��ٶȣ�ת����Ѫ�������١�
���岨��PW��������ʽ��PW̽ͷΪ����Ƭ̽ͷ��̽ͷ��ʱ����������ͳ������ա�PW��ʽ�£�̽ͷ��һ��ʱ�����ظ��ķ���һ�鳬��������ÿһ�鳬���������������̽ͷ�ɳ����������л�Ϊ�����ز����������Գ������䲨���н��ա�����̽ͷ���ջز�Ϊȫ��ȵĻز��źţ�����ض���ȵĻز��źž���Ҫ���루��ȣ�ѡͨ�Իز��źŽ��н�ȡ���乤��ԭ����ͼ3��ʾ��
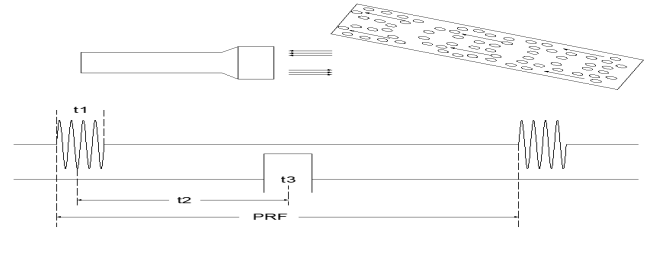
ͼ3�����岨ģʽ����ԭ��ͼ
����t1������������������t2������ѡͨ��t3��ȡ���ݻ���PRF���ظ�Ƶ��
��������CW��������ʽ:CW̽ͷΪ˫��Ƭ̽ͷ��̽ͷ������Ƭһ����Ϊ��������������һ����Ϊ������������������Ƭ�ֱ�ϵضԳ����źŽ�����������ͽ��ա���ˣ���������ͨ��ʱ������ض���ȵ�Ѫ���źţ�����ѡͨ������
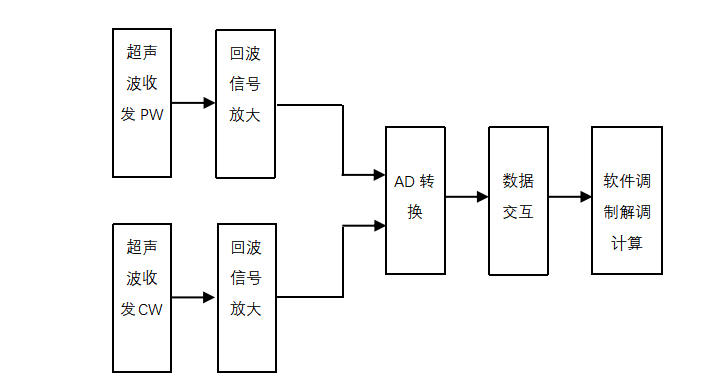
������������Ѫ�������ǹ���ԭ������ͼ4����ģ���·Ϊ������ͼ5�������ֵ�·Ϊ������ʾ��
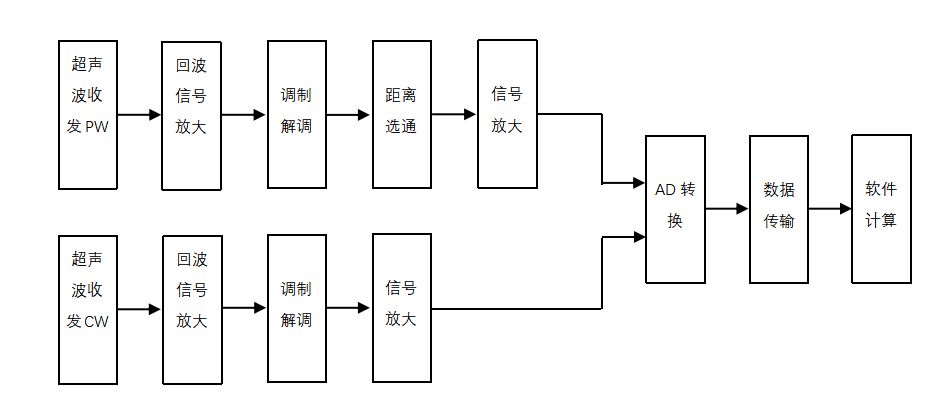
ͼ4����ģ���·Ϊ��������ԭ��ͼ
![]()
![]() ͼ5�������ֵ�·Ϊ��������ԭ��ͼ
ͼ5�������ֵ�·Ϊ��������ԭ��ͼ
5.��Ʒ�����÷�Χ/Ԥ����;/����֤
��1�����÷�Χ�����ھ�������������Ѫ�ܵ�Ѫ��������
��2��������Ⱥ�������ڸ�����ε���Ⱥ������������̥����
��3��Ԥ��ʹ�û�����������������Ѫ�������Dz�ƷӦ��ҽ�ƻ���ʹ�ã�ע��������Ӧ���ݲ�Ʒ������������ʹ�û�������������Ӧ�����¶ȡ�ʪ�ȡ���Դ���������ݡ�
��4������֤��Ӧ��ȷ��Ʒ�п��ܴ��ڵĽ���֤��
������Ʒ�Ľṹ�����ܲ�����ͬ��������Ԥ����;��Ϊ��ע�����г���������������Ѫ�������ǵ�ͨ�������������Ӧ��ϲ�Ʒʵ��������������ε������������ͬ�ͺš�����Ʒ���ٴ�Ӧ�ò���ͬ����Ӧ�ֱ����˵����
6.��Ʒ�IJ����¼���ʷ��¼
�������ڷ��շ���ʱӦ��עͬƷ��ҽ����е��Ʒ�IJ����¼���ʷ��¼��
����ʳƷҩƷ�����ֹ��ڳ���������Ѫ�������Dz����¼����������ͨ��MAUDE���ݿ������δ��������һ�����Ʒ����صIJ����¼���¼����������ҩƷ����������վ��ҽ����е�����¼���Ϣͨ���;����Ѷ��δ��������Ʒ����ز����¼���¼��
��һ�����ٴ�����
1.��Ʒ����Ҫ����
��Ҫ�ο�YY/T0316����ҽ����е ���չ�����ҽ����е��Ӧ�á��������չ����Ҫ�ᴩ��Ʒ��ơ����������к�ʹ�ü���Ʒ�����������������ڡ�Ҫ����ע�������˷��չ�����ƻ��������ԣ��������й����ķ��շ��������۹��̡���������ǰ���չ�������δ��֪�ķ��գ�Ӧ�����к�չ��Ϣ�ռ���һ�������쳣��ʱ���з������ۣ���ȡ���ƴ�ʩ�����·��չ����ļ���
��2����3����YY/T 0316����ҽ����е ���չ�����ҽ����е��Ӧ�á����ĸ�¼E ��ʾ���о��˳�����������Ѫ�������ǿ��ܴ���Σ�գ�Դ���ij�ʼ�¼��ͻ�����ʾ���Եظ�����Σ�գ�Դ�����ࡢΣ�գ�Դ���γɵ����ء����ܵĺ��֮��Ĺ�ϵ���������Ա������ʾ���ο���
���ڳ�����������Ѫ�������ǵ�ԭ�������ܺͽṹ�IJ��죬���¸����ķ���Ҫ�ؼ���ʾ���dz����Ķ�����ȫ���ġ���������ֻ�Ƿ��չ������̵���ɲ��֣����Ƿ��չ�����ȫ����ע��������Ӧ����YY/T 0316����ҽ����е ���չ�����ҽ����е��Ӧ�á����й涨�Ĺ��̺ͷ������ڲ�Ʒ�������������ڽ������γ��ļ�������һ�������Ĺ��̣������ж���ҽ����е�йص�Σ�գ�Դ�������ƺ�������صķ��ա�������Щ���ղ������������Ƶ���Ч�ԣ��Գ�ֱ�֤��Ʒ�İ�ȫ����Ч��
��2 ��Ʒ��Ҫ��ʼΣ�գ�Դ������
|
ͨ����� |
��ʼ�¼��ͻ���ʾ�� |
|
��������Ҫ�� |
��Ʋ����IJ��淶���ɴ����Ľ������֡���ǡ�Ӧ�ò��ֵ�����粿�ָ��뱣�������ȱ�ݣ����µ��Σ�շ��������ϵͣ����ܶ�ʹ��������ɵ��Σ�գ�Դ�����豸���������������ƶ�ʽ�豸�ȶ��Բ�豸�桢�ǡ��ߴֲڣ���ʹ������ɻ�е���ˣ���ż����Բ�����Ҫ�����豸�������ܽ��ͻ���������豸�������������ܳ������������㣬���µ��Σ�գ�Դ���ȡ� ���ܲ�����ǡ�������ٲ�����Χ���������롢����ѡͨ���Ȳ�����Ҫ�� ������ֱ�ӽӴ��������ϵ����������ԺͰ�ȫ�����⡣ ˵�����������Ϣ��ǡ�������淶��ʹ��˵����δ���豸��ʹ�á��豸��ά��������ʽ������Ƶ�ν�����ȷ��˵���������豸��������ʹ�õȡ� Ԫ�������������������ʧЧ����������쳣���ͷ����װ�ù��ϣ������豸�����趨����������������������ȫ���ܳ��������ȡ� �����Ľ�����ʹ������ʶ��ȷ�������ɶ�����ʹ�ȶ��Ե�����ָ�꽵�ͣ���ȫ���ܳ��������ȡ� ��Ӧ֤������֤��ȱʧ���ҽ����Ա�ĸ�벻�����»������˵ȡ� |
|
������� |
������̸��Ŀ��Ʋ���֣����Ƴ�����δ����������ֵ���֤�������豸���ܲ���ָ�겻���ϱ�Ҫ�� ������̵Ŀ��Ʋ���֣��������̹ؼ�������Ƶ�δ���м�⣬���²������������ϸ� �����Ŀ��Ʋ���֣������Э������ѡ���������Э��δ������Ч�������飬���²��ϸ�������Э��Ͷ�������� |
|
��������� |
��ǡ���İ�װ����Ʒ�������������豸����������� ���ʵ��Ļ����������ڳ����豸�涨�����ػ������¶ȡ�ʪ�ȡ�����ѹ���������豸�������豸�������������� |
|
�������� |
����ѧ���أ����¶ȡ�ʪ�ȣ������ȡ����䡢��ʪ�Ļ������ܵ����豸�������������ȡ� ��ų����أ���Ե�Ÿ��ŵ����жȣ�������Ÿ���������������ض��������豸�����������ȡ� ���ʵ���������Ӧ���豸�Ĺ����ѹ���ȶ��������豸�������������������������ȷ�ȡ� |
|
��ࡢ���� |
ʹ��˵�������Ƽ�����ϴ��������δ��ȷ�ϡ� ʹ����δ��Ҫ����з�������ϴ���������磺ʹ�ô�������������������δ����ȷ�ϣ� |
|
���úͷ��� |
δ�ṩ��Ϣ���ṩ��Ϣ����֣�δ��ʹ��˵�����ж��豸�ķ����ﴦ�ý�����ʾ��˵���� |
|
���� |
���������ԣ�������Ӵ��IJ�������ѡ�����¹����ȷ�Ӧ�� |
|
������ |
���ȱ���������ܵ�ʹ�ô��� �����Ļ�ȱ��ʹ��˵���飺��ȱ����ϸ��ʹ�÷�����ȱ�ٱ�Ҫ�ļ���������ȱ�ٱ�Ҫ�ľ���˵����ȱ�ٱ�Ҫ�ĵ�·ͼ��Ԫ�����嵥��ȱ����������滷�����������ƣ��豸�ڹ���״̬����Ͽ������ӵ��ߡ��豸��Ԫ�������ֹ��ϣ������пɲ���Σ�վ�ʾ���㣻ʹ��ǰδ����豸����״̬������˵�����ڸ��ӡ�������δ˵�������ȷά���������豸/����������ͻ����а�װ��δ��ϸ˵��װ����̺�ע�����δ˵�������ų�ָ�ϡ� ��ϴ��������������ȷ�� ���߲������˶��� ��ȱ�������ġ�δ����ѵ����Աʹ�ã�ʹ����������δ����ѵ����ѵ���㣬������ȷʹ�ú�ά���������豸���豸/����������ʹ�á� |
|
ʧЧģʽ |
�������ϣ��û��ĵ����ᵽ�Ĺ��ܲ���ִ�С� |
��3 Σ�գ�Դ�����ࡢΣ�գ�Դ���γɵ����ء����ܵĺ��֮��Ĺ�ϵ
|
Σ�գ�Դ������ |
Σ�գ�Դ���γɵ����� |
���ܵĺ�� |
|
|
���� Σ�գ�Դ�� |
����� |
���ܹ�ͬʹ�õ��豸�����������ӡ�����ƶ��绰�ȣ��Գ�����������Ѫ�������ǵĵ�Ÿ��ţ�����ŵ�Գ�����������Ѫ�������Dz����ĸ��ţ�������������Ѫ�������Dz����ĵ�ų��Կ��ܹ�ͬʹ�õ��豸��Ӱ�졣 |
Ƶ���쳣Ӱ����� |
|
���� |
Ӧ�ò���©����������Ҫ��ԵʧЧ���ӵز������Ե��迹��Ӧ�ò�������粿��û�г�ָ��룻�豸�ĵ�Դ��ͷʣ���ѹ���ߣ�������ǵķ����ַ�ղ����� |
ʹ�����ߵ�����ˡ������� |
|
|
���� |
Ӧ�ò�����ЧӦ���۵��¹��� |
����������� |
|
|
���ܰ�ȫ |
�豸���ϻ�ʧ�أ����¹������������������壻 ��Ʒ��������ơ���ʾ����ʧЧ����ϡ� |
�������������֯ϸ�����ˡ� |
|
|
��� |
�豸���������������ƶ�ʽ�豸�ȶ��Բ�����㵹�� |
����е���ˡ� |
|
|
����ѧΣ�գ�Դ�� |
�ٴλ��Ⱦ |
������Ӵ��IJ���δ����ϴ����������Ľ����Ⱦ�� |
��������߽Ӵ����½����Ⱦ�� |
|
ԭ���� |
������������Ѫ��������������Ӵ����ֵ�ԭ�����ж��к���������ɵ�Σ���� |
���������DZ�ڵ�Σ���� |
|
|
��ѧΣ�գ�Դ�� |
������������ |
ʹ�õ�����������������������Σ���� |
�����������DZ�ڵ�Σ���� |
|
��Ϣ Σ�գ�Դ�� |
��� |
�������ȱ�ٻ���ȷ����ǵ�λ�ò���ȷ�������������κ�������ϵȡ� |
�豸��״̬�����������Ϣ������ |
|
����˵���� |
˵����δ�Բ���/������װ��ʹ������˵����˵����δ�������������ά����Ϣ������ϸ˵����˵����Բ�Ʒ�������������÷�Χ��ʹ�����Ƶ��������淶����������˵����δ�Թ����Ų�����ϸ˵����˵����δ�Ժ�����Ԥ�������ý��о��档 |
��Ԥ�ڻ�Χʹ�ã��豸�������������������������ƫ����豸��ʹ�����ܵ������˺��� |
|
|
���� Σ�գ�Դ�� |
ʹ�� ���� |
��ע�������˹涨��ʹ�û���������ʹ�ò�Ʒ�� |
���豸����Ʒ�������ͣ�����ʱ����ʹ�����ܵ������˺��� |
|
ʧЧ������Σ�գ�Դ�� |
������������ |
��ע�������˹涨�����滷�������������Ʒ�� |
������ɲ�Ʒ������������������Ʒ�������͡� |
|
ʧЧģʽ |
Ԫ�������ϡ�������ƴ���©�� |
Ѫ��������ȷ |
|
2.��Ʒ����Ҫ�����Ҫ����ָ��
�걨��Ʒ�����ܲ�������Ӧ�������Ʒ���÷�Χ��Ҫ�����������Ҫ���ǵIJ�Ʒ��Ҫ����ָ�꣬���и��ӹ��ܣ�ע��������Ӧ����������롢���������ڸ��ӹ����Ĺ��ұ�����ҵ����ע���������粻������������������ұ�����ҵ��Ҫ��Ӧ��˵�����ɡ�
2.1����
2.1.1������������
Ӧ�涨��Ʒ�����������������¶ȡ�ʪ�ȡ�����ѹ������Դ�����ȣ���
2.1.2������������Ѫ�������Dzο�ִ��YY/T 0593����������������Ѫ�������ǡ�����Ҫ��
2.1.3Ӧ�涨��ҵ������ļ��������ĸ���ܣ�ͬʱ���㡶ҽ����е����ע�Ἴ�����ָ��ԭ��Ҫ��
2.1.4 ��̤���أ������ã�
Ӧ����YY 1057����ҽ�ý�̤����ͨ�ü�������������Ҫ��
2.1.5�ͷ�ף������ã�
Ӧ˵������ڷ�ʽ���ߴ�ȡ����ھ����Զ����ڹ��ܵģ�Ӧ�оٹ����嵥����ȷ����ָ�꣬���Զ��н��ĵ��������Զ������ķ�Χ�ȡ�
2.1.6 ���Ʒ�����ƶ�ҽ����е��Ӧ���ϡ��ƶ�ҽ����еע�Ἴ�����ָ��ԭ�����Ҫ��
2.1.7 ���Ʒ�����������ӹ��ܻ���ô洢ý��ķ�ʽ���Խ��е������ݽ�����Զ�̿��Ƶģ�Ӧ���ϡ�ҽ����е���簲ȫע�Ἴ�����ָ��ԭ�����Ҫ��
2.2��ȫҪ��
2.2.1������ȫҪ��
Ӧ����GB 9706.1����ҽ�õ����豸��1���֣���ȫͨ��Ҫ����Ҫ��
�����ã�Ӧ����GB 9706.15����ҽ�õ����豸��1���֣���ȫͨ��Ҫ����Ҫ��
Ӧ����GB 9706.9����ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸ר�ð�ȫҪ����Ҫ��
2.2.2��ż�����Ҫ��
Ӧ����GB 9706.9����ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸ר�ð�ȫҪ���е�36�¼�YY 0505����ҽ�õ����豸 ��1-2���֣���ȫͨ��Ҫ�� ���б�����ż��� Ҫ������顷���й涨��Ҫ��
3.��Ʒ���о�Ҫ��
3.1��Ʒ�����о�
3.1.1�ṩ��Ʒ����Ҫ��������ָ���ȷ�����ݣ�����ָ�꽨�����YY/T 0593����������������Ѫ�������ǡ�������Ӧ����������õ�����Ӧ������˵�������ڲ����õ�������鷽��Ӧ��ȷ�����õĺ������ɡ�
3.1.2 Ӧ�Բ�Ʒ˵���������Ƶ���Ҫ���ܵ�ʵ��ԭ��������������Ҫʱ���Ը�ͼ˵���������ٴ����������������������ԭ�����ٴ����ܵ��㷨ʵ��Ӧ����˵����
�����ƾ��ж���ȵIJ�Ʒ��Ӧ˵�������ʵ�ֵķ�ʽ����Ҫע�⣬����ȱ����Ǵ�һ��̽ͷͨ��ͬʱ��ö����ȵĶ�����Ƶ���źţ�����һ��̽ͷͨ����ʱ�ε��λ��һ����ȵĶ�����Ƶ���ź���ͬʱ��ʾ�������ź�Ƶ�IJ�Ʒ�ǵ���ȵIJ�Ʒ����Ʒ����Ҫ��Ʒ˵�����ж���ͨ������������ı���Ӧ��ʵ��һ�¡�
�ṩ���ӹ��ܣ���Ӱ���ܵȣ����ڲ���֤���ϣ��磺�ٴ����塢ʵ�ַ���������Ҫ���ӹ��ܶԾ�������������Ѫ�ܲ�����Ƶ�ף���Ӱ��ȡ�
�ṩ˨�Ӽ���ԭ����ʵ�ַ�ʽ��
3.1.3�ṩ������ģ�������Ϣ���о����ϡ�ֱ�Ӳɹ���Ʒ��ģ�ģ��ṩ��ģ�����̡���ģ��;�����������������ϡ�������ģ�ģ��ṩ��ģ����֤���ϵȡ�
3.1.4����̽ͷ�ij���Ƶ�ʵ�ѡ��ֱ��Ӱ�������Ƶ���źŵIJɼ������������������Ǻ���֯�Ĵ��Ժ�������ȣ�������������Ѫ�������Dz��õĵ���Ƶ��ΪYY/T 0593����������������Ѫ�������ǡ������Ƽ���Ƶ�ʣ����ڲ��÷ǵ���Ƶ�ʵ�̽ͷ��Ӧ˵��ʹ�õ����ɺ�ʵ��ԭ����
3.1.5���� GB 9706.237����ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸�Ļ�����ȫ�ͻ�������ר��Ҫ�������ƾ���TI����ָ�����Զ����ƹ��ܵIJ�Ʒ��Ӧ�߱���ʾʵʱ��������ʣ�mW���Ĺ��ܣ�˵����ʵ�ֵķ�ʽ���ύ��֤���ϡ�
3.1.6��Ʒ���мͷ���볬��̽ͷ���ϵͳ�ģ�Ӧ˵���书��ʵ�����ֶ������Զ��ġ������ֶ����ڵ����ϵͳ��Ӧ�ṩ�ͷ�ṹͼ��ϵͳʹ��˵�������ھ����Զ����ܵ����ϵͳ�����Զ�ɨ�衢����������Ѫ�ܣ�ѡ�����Ѫ�ܣ��Զ��н��ȹ��ܣ�Ӧ˵�������Զ����ܼ���ʵ�ַ�ʽ�������Զ����ܵĿɿ��Բ��ṩ��֤���ϡ�
���ƾ����Զ��н����ܵ����ϵͳ��Ӧ����ѹ�����л���λ��ֹ�ȷ�ֹ�����˺��Ĺ��ܣ�˵����ʵ�ַ�ʽ���ṩ��Ӧ����֤���ϡ�
3.2���������������о�
���ڳ�����������Ѫ�������ǣ�������Ӵ��IJ���һ�����������������̽ͷ���ͷ�ȡ�
Ӧ����GB/T 16886ϵ�б������������������ۣ�Ӧ�ṩ�Ӵ��������ơ��������ϡ��Ӵ����ʣ��Ӵ����͡��Ӵ�ʱ�䣩������������ѧ���飬���ٰ������·����Ҫ��ϸ�����ԡ��ٷ��ͳ�����Ӧ���̼�������������ѧ���飬Ӧ��֤�������ɡ�
3.3�������������
Ӧ�涨��������ࡢ�������գ���Ӧ���ݳ���̽ͷ���ٴ�ʹ������ṩ̽ͷ����ࡢ������������յ��о����ϡ�
�糬��̽ͷԤ�ڽ��Ӵ�����Ƥ����һ������ն��û������ķ�������ȷ�Ƽ����������գ������Ͳ������Լ����Ƽ���������ȷ�������ݡ�
�糬��̽ͷ�����ն��û�����ķ�����Ӧ����ȷ�Ƽ���������գ������Ͳ����������Ƽ����������ȷ�������ݡ��Կ��������λ�������IJ�Ʒ��Ӧ���ṩ��Ʒ�Ƽ���������������Ե�����о����ϡ��糬��̽ͷ��һ����������ʽ�ṩ��Ӧ����ȷ������գ������Ͳ�����������֤ˮƽ��SAL�������ṩ���ȷ�ϱ��档
�����ʹ�õķ������׳��ֲ�����Ӧ����ȷ��������Ϣ����ȡ�Ĵ������������ṩ�о����ϡ�
3.4��Ʒʹ�����Ͱ�װ�о�
ע��������Ӧ�ṩ���ϡ���Դҽ����еʹ������ע�Ἴ�����ָ��ԭ�IJ�Ʒʹ���������۱��档Ӧ���ڷ��շ����ص㿼��Ԫ�����������ϻ���ʹ�û���������ʪ�ȣ��Բ�Ʒ���ա������Ӱ�졣����̽ͷ��Ϊ�IJ�ʹ��ʱ��Ӧ�������ۡ�
Ӧ�Բ�Ʒ�İ�װ����װ�������ṩ�о����ϣ������������Ч���ǶԲ�Ʒ�����������������������ܱ��ֹ��������Ҳ�Ʒ��װ������
��Ʒ��װ���Ӧ����GB/T191������װ����ͼʾ��־������YY/T 0466.1����ҽ����е����ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���֣�ͨ��Ҫ����Ҫ���ṩ����֤�ݺ�ʹ�������������Ե����ݡ�
3.5�����о�
��ijЩ��������⣬������������Ѫ��������ͨ����������������������豸��������Ӧ���ա�ҽ����е����ע�Ἴ�����ָ��ԭ������Ҫ���ṩһ�ݲ�Ʒ�����������ĵ������Ʒ�����������ӹ��ܻ���ô洢ý��ķ�ʽ���Խ��е������ݽ�����Զ�̿��Ƶģ���Ҫ���ա�ҽ����е���簲ȫע�Ἴ�����ָ��ԭ�����ύһ�����簲ȫ�����ĵ������Ʒ�����ƶ�ҽ���豸����Ӧ����ϡ��ƶ�ҽ����еע�Ἴ�����ָ��ԭ��Ҫ���ύ��Ӧע���걨���ϡ�
3.6���ܰ�ȫ
������GB 9706.9����ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸��ȫר��Ҫ���Ļ����ϣ���Ӧ�涨�����������ֵ����ȷ���䰲ȫ�ԡ�Ӧ�����������ֵ���õĺ����Խ��з�������ȷ�趨�����ݣ����ύ�豸ʵ����������ܹ�������ֵҪ�����֤���档��ֵ���趨�����Եķ���Ӧ�ο�ҵ��ͨ�õ���
3.7��������Ҫ��
��ҵӦ������GB/T 14710����ҽ�õ�������Ҫ�����鷽��������YY/T 1420����ҽ�ó����豸����Ҫ�����鷽��������Ҫ���ṩ�����������֤���ϣ�����������Ŀ������Ҫ��Ͳ�����ĿӦ���ϱ���Ҫ��
4.ͬһע�ᵥԪ��ע���������Բ�Ʒȷ��ԭ���ʵ��
4.1���Ͳ�ƷӦ��ͬһע�ᵥԪ���ܹ���������Ԫ��������Ʒ��ȫ�Ժ���Ч�ԵIJ�Ʒ��
4.2Ӧ���ǹ�������ȫ���ṹ��ӡ�������ߵIJ�Ʒ��
4.3���Ͳ�Ʒ�뱻������Ʒ�ĵ�Դ���Ӧ��ȫ��ͬ����������Ʒ�Ĺ��ܡ�����̽ͷ�����������ͺ�ӦΪ�����ͺŵ��Ӽ�����Ҫ����ָ��Ӧ������ͺ������
4.4ע�ᵥԪ�ڸ����ͺŲ�Ʒ����Ҫ��ȫָ�ꡢ����ָ�겻�ܱ�ijһ�ͺŲ�Ʒȫ������ʱ����Ӧѡ�ǰ�ȫָ�ꡢ����ָ�������ͺ���Ϊ���Ͳ�Ʒ��ͬʱ��Ӧ����������Ʒ��δ�������ͺ������ǵİ�ȫָ�꼰����ָ�ꡣ
4.5��û�г���֤���ܹ�֤��ͬһע�ᵥԪ�ڲ�ͬ�ͺŹ���Ʒ֮���ż������ܿ��Ը���ʱ��Ӧѡȡÿһ�ͺŹ���Ʒ���е�ż�����Ŀ��⡣
��һ���ٴ���������
�걨��Ʒ�����÷�Χ��Ӧ�����ٴ��������������۵ķ�Χ��
1. ���Ʒ���ϡ����ڽ����ٴ�����ҽ����еĿ¼�����������������ύ�ٴ��������ϡ����Ʒ���и������ܣ��綳�ᡢ¼��Mģ��˨�Ӽ��ȣ����ڡ����ڽ����ٴ�����ҽ����еĿ¼���ڵIJ�Ʒ��
2. ���Ʒ���������ڽ����ٴ�����ҽ����еĿ¼�������������Ʒ���з�����Ϲ��ܻ�������̽ͷ�ȣ���ɰ��չ���ҩ��ַ����ġ�ҽ����е�ٴ����ۼ���ָ��ԭ���������Ƿ�չҽ����е�ٴ����鼼��ָ��ԭ����ҽ����е�ٴ����۵�ͬ����֤����ָ��ԭ����ҽ����еע���걨�ٴ����۱��漼��ָ��ԭ������ط����ļ��ύ�ٴ��������ϣ���֤����Ʒ�İ�ȫ�Ժ���Ч�ԡ�
��������Ʒ˵����ͱ�ǩ����
��Ʒ˵����ͱ�ǩ�ı�дӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨������ر��Ĺ涨��һ��Ӧ��������Ҫ��
1.˵����
˵����Ӧ����������࣬Ӧʹ����������������ļ���ṹ�����������Ķ�������ʹ�÷��Ż�ͼʾ��
ÿ̨�豸��Ӧ����˵���飬˵����Ӧ������ҽ����е˵����ͱ�ǩ�����涨������ر��涨��һ��Ӧ�����������ݣ�
1.1��Ʒ���ƣ���ȷ��Ʒ���ơ���Ʒ�ͺš��������������塣
1.2����ע���˵����ơ�ס������ϵ��ʽ���ۺ����λ��
1.3����������ҵ�����ơ�ס����������ַ����ϵ��ʽ����������֤���ţ�ί�������Ļ�Ӧ����ע������ҵ�����ơ�ס����������ַ����������֤��š�
1.4����ҽ����еע��֤��ż���Ʒ����Ҫ���š�
1.5��Ʒ���ܣ�������Ʒ����Ҫ����顣
1.6��Ҫ�ṹ��ɣ�ע��������Ӧ�涨����Ʒ�Ľṹ�����Ӧ�в�Ʒ�������ͼʾ��˵����
1.7��Ʒ���÷�Χ������֤�������ٴ��������ϼ��������������
1.8ע�������ʾ����ʾ���ݣ�Ӧ���ա�ҽ����е˵����ͱ�ǩ�����涨���е�ʮһ����Ҫ�������飻��е��������������λ����ע�⣨����ʱ������е�ڷ�������ʱ�ľ���˵����Ӧ����ע�����ڵ�����װ�����ʶ���ɵ�Σ�գ�Ӧ����������������Ѫ���������������豸��DZ�ڵĵ�Ÿ��Ż��������ŵ������Ϣ���Լ��йر�����Щ���ŵĽ��顣
1.9��װ��ʹ��˵����ע��������Ӧ��ȷ��Ʒ��ʹ�÷�����Ӧ��ȷ��Ҫ�û����а�װ���֣���ɲ�ж������İ�װ�����Է�������ע�����Ӧ��ȷ����ͣ�ú��ʹ��ǰ���ͼ�����
1.10������ά��������ע��������Ӧ������Ʒά���ͱ��������ڼ��ķ��������п����û������ų��Ĺ��ϣ���Ӧ˵�����ϵ�����Ͳ�����ԭ���ų������ȡ�
1.11����������ע��������Ӧ���ݲ�Ʒ���ԣ���ȷ���䷽����������
1.12����������ע��������Ӧ���ݲ�Ʒ���ԣ���ȷ���滷��Ҫ��
1.13Ӧ��ȷ�������ڡ�ʹ��������Ԥ��ʹ�ü�ά�������µĶ��ڼ��ʱ�䡣
1.14Ӧ��ȷ��Ʒ����嵥���������������Ʒ�����Ʒ�������ڼ�����������˵����
1.15Ӧ������ع��ұ�����ҵ���еĹ涨��������Ʒ��ǩ���õ�ͼ�Ρ����š���д�����ݵĽ��͡�
1.16������������������ע��������Ӧ�������Ʒ����г���Ʒ���������������ķ�����
1.17��ȷ˵����ı��ƺ������ڡ�
1.18����GB 9706.1����ҽ�õ����豸��1���֣���ȫͨ��Ҫ����GB 9706.15����ҽ�õ����豸 ��1-1���֣�ͨ�ð�ȫҪ�� ���б���ҽ�õ���ϵͳ��ȫҪ����GB 9706.9����ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸ר�ð�ȫҪ����Ҫ���ṩ��Ӧ��Ϣ��
1.19����YY0505����ҽ�õ����豸 ��1-2���֣���ȫͨ��Ҫ�� ���б� ��ż��� Ҫ������顷����Ҫ��������ϵ�ż����Է���Ҫ���������
1.20����YY/T 0593����������������Ѫ�������ǡ����е�Ҫ���ṩ�����Ϣ��
1.21Ӧ��˵��������ȷ���������汾��Ӧ��������Ҫ�����ͼʾ��ʹ�ò���˵����
1.22����صIJ�ƷӦ˵��ʹ��ʱ����ά����ʽ��
��Ʒ˵��������ݾ�Ӧ����ȷ����Դ�����������ϡ��о����ϵ�ע���걨���ϵ����ݱ���һ�¡�˵�������漰����������ǰ��ע���걨������δ�����ģ������ύ��Ӧ��֤���ϡ�
2.��ǩ
������������Ѫ�������ǵı�ǩӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨����YY/T 0466.1����ҽ����е������ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���� ͨ��Ҫ��������ر���Ҫ��
������������Ѫ�������ǵı�ǩ��λ�û��ߴ�С������ȫ�������������ݵģ�����Ӧ����ע��Ʒ���ơ��ͺš�����������ں�ʹ��������ʧЧ���ڣ����ڱ�ǩ����ȷ�������������˵����������ʹ�õķ���û�����еı���Ӧ���ڳ�����������Ѫ�������ǵ�����ļ��ж���Щ���Ž���˵����
����������������ϵ�ļ�
1.�������չ��̼����̿��Ƶ�
ע��������Ӧ�����걨��Ʒ��ʵ�������������ͼ����ʽ���������չ��̽�����ϸ����������������ͼ��һ�������еĹ��̿��Ƶ㡣��������ͼ�еĹؼ�������������Ӧ������ͼ�α�ʾ��
������������Ѫ�������Dz�Ʒ���վ���˵����������������Ѫ�������Dz�Ʒ��������һ��������ӡ����ԡ���װ��������¼���ϻ��ȹ���
ע����˵����Ϊ������˵����ע�������˿ɸ��ݲ�Ʒ���������Ʒ�������պ��̿��Ƶ㡣
2.���ơ����������������
ע��������Ӧ�������걨��Ʒ�йص����Ƴ��غ���������������и�������Ҫ�����������ݣ�
���Ƴ��أ���ַ��λ�á���������ƻ��������������豸����֤�豸�ȡ�
�������أ���ַ��λ�á�������������������������豸������װ�������ӺͲ���װ�õȡ�
���������
[1] GB 9706.1��ҽ�õ����豸 ��1���֣���ȫͨ��Ҫ��[S].
[2]GB 9706.15��ҽ�õ����豸 ��1-1���֣�ͨ�ð�ȫҪ�� ���б���ҽ�õ���ϵͳ��ȫҪ��[S].
[3]GB 9706.9��ҽ�õ����豸��2-37���֣�������Ϻͼ�
���豸ר�ð�ȫҪ��[S].
[4] GB 9706.237��ҽ�õ����豸 ��2-37���֣�������Ϻͼ�豸�Ļ�����ȫ�ͻ�������ר��Ҫ��[S].
[5] GB/T 14710��ҽ�õ�������Ҫ�����鷽��[S].
[6] GB/T 16886.1��ҽ����е����ѧ���� ��1���֣����չ��������е�����������[S].
[7] GB/T 16886.5��ҽ����е����ѧ���� ��5���֣�����ϸ����������[S].
[8] GB/T 16886.10��ҽ����е����ѧ���� ��10���֣��̼���Ƥ����������[S].
[9] YY 0505��ҽ�õ����豸 ��1-2���֣���ȫͨ��Ҫ�� ���б�����ż��� Ҫ�������[S].
[10] YY /T 0593��������������Ѫ��������[S].
[11] YY/T 0458�����������շ�Ѫ����ģ�ļ���Ҫ��[S].
[12] YY/T 0704������������������ϵͳ�������鷽��[S].
[13] YY/T 0705����������������ϵͳ���鷽��[S].
[14] YY/T 1142��ҽ�ó����豸��̽ͷƵ�����ԵIJ��Է���[S].
[15] YY/T 1420��ҽ�ó����豸����Ҫ�����鷽��[S].
[16] YY/T 0466.1��ҽ����е ����ҽ����е��ǩ����Ǻ��ṩ��Ϣ�ķ��� ��1���֣�ͨ��Ҫ��[S].
ע�����ϱ��������°汾��
�ġ����Ƶ�λ
����ʡҩƷ�ල��������������