

| �����������HSV�������⼰�����Լ�ע�����ָ��ԭ��ָ��ԭ��ּ��ָ��ע�������˶Ե�������������⼰�����Լ�ע���걨���ϵ�����д��ͬʱҲΪ�������������ṩ�ο�����ָ��ԭ���ǶԵ���������� |
�����������HSV�������⼰�����Լ�
ע�����ָ��ԭ��
��ָ��ԭ��ּ��ָ��ע�������˶Ե�������������⼰�����Լ�ע���걨���ϵ�����д��ͬʱҲΪ�������������ṩ�ο���
��ָ��ԭ���Ƕ���������������⼰�����Լ���һ��Ҫ��ע��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ��������������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ����
��ָ��ԭ������ע�������˺ͼ���������Աʹ����ָ�����ļ����������������������漰���������������Ϊ����ǿ��ִ�У�Ӧ����ѭ��ط����ǰ����ʹ�ñ�ָ��ԭ��������ܹ�������ط���Ҫ�������������Ҳ���Բ��ã�������Ҫ�ṩ��ϸ���о�����֤���ϡ�
��ָ��ԭ���������з���ͱ���ϵ�Լ���ǰ��֪ˮƽ���ƶ������ŷ���ͱ��IJ����������Լ���ѧ�����IJ��Ϸ�չ���������Ҳ����ʱ���е�����
һ�����÷�Χ
��ָ��ԭ����������������ʵʱӫ��PCR��Real-Time PCR���������⼼�������ض������������1�ͣ�Herpes simplex virus type 1��HSV-1����/�������2�ͣ�Herpes simplex virus type 2��HSV-2����������Ϊ���Ŀ�꣬����Դ�����������е����������DNA�����������Լ�����ͼ����Լ����ٴ���ҪӦ������ֳ�������е����������Ⱦ��������ϡ���Ӧ����������λ��Ӧ������ƺ������ٴ����������֤��
�ٴ����ڼ������������ļ���Լ����ɸ��ݲ�ͬ����Ӧ֤ѡ���������������ͣ���ֳ���������Ҫ��������Ϊ��ֳ��Ƥ���ĤƤ��걾������������Һ��������Һ�ȣ�����������ֳ�����ӣ���Ů�ԵĹ������ӻ��������ӡ����Ե�������ӣ�����Ӧ����������ϵͳ��Ⱦʱ�������漰�Լ�Һ������
���ڲ�����������ѧ����������������������������ȣ������߲��������������������Լ�Һ�ȣ���HSV�����������Լ������ܲ���Ҫ����ȫ���û���������������ȫ�棬ע��������Ӧ���ձ�ָ��ԭ�����ݲ�Ʒ���Զ����ò��ֽ������ۣ����������������ۡ�
��ָ��ԭ��������HSV�����⼰�����Լ����в�Ʒע��ͱ��ע������Ρ���ָ��ԭ�����ע���걨�����еIJ������ݽ���д������δ������Ӧ�����ϡ����ڹ�����������Լ�ע���걨����Ҫ�����֤���ļ���ʽ�Ĺ��桷����ط���Ҫ��ͬʱ����ο������Լ���Լ�������������ע�����ָ��ԭ�����õļ����ļ�Ҫ����
����ע�����Ҫ��
��һ�������Ϣ
1.��Ʒ���Ƽ��������
��Ʒ����Ӧ���ϡ���������Լ�ע���뱸�������취������ط����Ҫ���絥�������1�ͣ�HSV-1���������Լ��У�PCR-ӫ��̽�뷨�������������2�ͣ�HSV-2���������Լ��У�PCR-ӫ��̽�뷨���ȡ����ա���������Լ������������Ʒ���յ�������������Լ��������������Ϊ6840��
2.ע�������������ύ��Ʒ�б��������ļ����걨ǰ���ܻ�������ϵ�����ͨ��¼���������������ļ���
��������������
����������Ҫ������������Ʒ������Ԥ����;���걨��Ʒ������ʷ��������˵�������ݡ����У���ע���������ݣ�
1. ����ԭ��Ӧ��ϸ˵����Ʒ�����õļ���ԭ����������̣���ȷ��Ʒ���İл������У���ѡ�����Ժ���������ԽϸߵĻ���Ͱ����У�ͬʱ��Ӧ��������Ч�ʡ��ṩ�����̽���������ݡ��ṩ������ȡ���ֹ����Զ���ȡ��ʽӦ�ֱ���ȷ����PCR������ʱ�䣬�ṩ��ͬ���û��͵ļ��ͨ���������ã���
2. Ԥ����;����ȷ�ɼ����ͱ𡢰л����������͡�������λ��������Ⱥ���ٴ���Ӧ֤�ȡ�
3. ��������ͬ���/��ǰ����Ʒ�ıȽϲ���Ӧ���ذ�������ѧ������ԭ�����������͡�������λ��������ʽ�����л�����ɳɷ֡��ڱꡢ�ʿ�Ʒ���ж�����ͬHSV���ͼ��������Ԥ����;��������Ⱥ���������ܺ��ٴ����ܵȷ��档
���������ٴ�����
1.��Ʒ����Ҫ���鱨��
���ա�ҽ����е��Ʒ����Ҫ���дָ��ԭ��Ҫ���д��Ʒ����Ҫ���ύ������ͬ���η��ϲ�Ʒ����Ҫ���ȫ��Ŀ���鱨�档�ύ����Ӧ���ϡ���������Լ�ע���걨����Ҫ��˵������ҽ����еע���Լ�����涨��������ļ���Ҫ��
2.���������о�
ע��������Ӧ����ԭ���Ϻ��������վ���ѡ���ȷ�ϡ�����������ϵ�õ���Ч���Ʋ��ұ�֤��Ʒ�����ȶ��Ļ����ϣ�����������ȷ���ļ��ϵͳ���з�������������
����ÿ��������ܵ����۶�Ӧ���������о�Ŀ�ġ��о����������鷽�����������ݡ�ͳ�Ʒ�������ϸ���ϡ��йط���������֤�ı�����ϢҲӦ���걨�������������֣���������ص㡢���õ��Լ����ơ��������ţ��������ƺ��ͺţ��������ͺ���Դ�ȡ������������������鷽�����Բο����ʻ�����й���������Լ�����������ָ��ԭ����С�
���������������������Ļ�����Ϣ������ȷ������������Դ���������͡������ɼ��ʹ�����ʽ��ϡ�ͷ�ʽ��Ũ�Ȼ�ζȣ���ֵ��ȷ�����̼����ݵȡ��о��в��õ�HSV-1��/��HSV-2����������Ӧ���ÿ�ѧ�����ķ���ȷ�����ͱ������Ժ�Ũ��ˮƽ���ύ������������ϡ������о�����Ũ����Դ�����ʱ�Ʒ��������������������һ��ӦΪ��ʵ����������������������ٲ����Ƚ��з����������������漰ϡ�ͺ��⣬Ӧ������������������һ�µ����Ի��ʽ���ϡ�͡����ڸ��������в��õ����������������������о������зֱ��ṩ������Ϣ�б�������Ͱ������о�����������Ӧ�������
������õIJ�ͬ�������ͷֱ���з��������о���Ӧ���������IJ�����λ��
���Լ����ڲ�ͬ���û��ͣ���Ҫ�ڲ�ͬ�����Ϸֱ�����������������걨��Ʒ������ͬ�İ�װ�����Ҫ�Ը���װ�����з�������֤��
�������ض����·������ܽ����о���
2.1�����ȶ���
Ӧ��ֿ���ʵ��ʹ�ù����������ɼ������������估����ȸ����ε��������Բ�ͬ�������͵������ȶ��Էֱ�����о������в�ͬ�������ɼ������������粻ͬ�������ӡ��������̡��ɼ���δ�����������������벻ͬ�ѽ�Һ/����Һ�������ȣ�����ͬ������������ʣ���������Һ����������������ͬ����������ͱ������������漰��������ֱ�����ȶ����о���һ�������������ʵ�����估���������µı���������֤�����䶳�����������Ӧ�Զ��ڴ������к�����֤��
�������ȡҺ�ɲ��������м�⣬����Ժ�����ȡҺ�ı��������ͱ���ʱ������о���
2.2���õ���������
��ϲ�Ʒ��������Ⱥ��Ԥ����;��HSV���ͣ�������Ʒ���õ��������͡�������λ�������ɼ������������ռ�/������ʣ������Ӽ������ú������������������ԡ�
���걨��Ʒ��������������ͣ�����Һ����������Һ���ӡ�����������ӡ�Ů�Թ���������������Ϊ��ͬ������������ֱ���з��������о����о�����Ӧ���������IJ�����λ��������ֳ�����ֱ��Ƥ��ճĤ��Ů����������������������ȣ������Ʒ�����������������ͣ�Ӧ�����������������ͽ���ȫ����������
����ò�ͬ�����ɼ�������ͬ��������Һ/ϡ��Һ ����ͬ���������ȣ���ֱ���з��������о���
�����ѻ�õ��������ͣ�����Һ���ɲ��ֲ������ٴ����Ի��ʾ���һ���Ե��˹��Ʊ����Ի��ʽ�����ͼ����ȷ����ע�����������ֿ��ǻ���ЧӦ��Ӱ�죬��֤�˹��Ʊ�������Ԥ�����������ļ����������ƫ����ύ�о����ϡ�
2.3��ҵ�ο�Ʒ��֤
������Ҫԭ�����о������е���ҵ�ο�Ʒ�����������������������Ʒ����ҵ�ο�Ʒ���м��鲢�ṩ��ϸ���������ݡ�
2.4ȷ��
�ɲ����걨�Լ�������ȷ�ͱ��Ũ���������������в�Ʒͬʱ����ٴ������Ƚϼ����֮���һ���Գ̶ȡ��о�Ӧ����һ�����������Ժ�������������ע�����һ�������������ж�ֵ�����������ͻ�ϸ�Ⱦ������
��ͬ����������Ӧ�ֱ����ȷ�����ۡ�
2.5���ܶ�
ע�������˸����Լ��м�ⷶΧѡ����Ӧ��HSV�ͱ���о��ܶ���֤������HSV-1��HSV-2ͨ�ü����ͼ����Լ���ע�������������HSV-1��HSV-2���б�ֱ���о��ܶ����ۣ���������������HSV-1��HSV-2�IJ�ͬˮƽ�����ڷ����Լ���ÿ��������ֱ��ṩHSV-1��HSV-2�ķ��������
ע��������Ӧ�Ծ��ܶ�ָ������۱���������Ҫ������Ӧ�ͱ�������Է����ʣ�Ctֵ��������ϵ���ķ�Χ�ȡ�Ӧ�Կ���Ӱ���⾫�ܶȵ���Ҫ������ƺ��������鷽�����о��ܶ����ۣ��������С�ʱ�䡢�����ߡ��������Լ����κ͵ص��Ӱ�쾫�ܶȵ������ȡ�ע��������Ӧ�Լ�����ݽ���ͳ�Ʒ���������ظ��ԡ�ʵ�����ھ��ܶȡ�ʵ���Ҽ侫�ܶȡ����侫�ܶȵȽ����
���ܶ���������Ӧ����������ȡ���衣Ӧ�趨�����ľ��ܶ��������ڣ�����Ϊ������20��ļ�⡣�ɲ��ÿ���Դ�IJ����ꡢ�ٴ�����������ٴ������������飬���ٲ���3��Ũ��ˮƽ������Ҫ�����£�
����������������Ũ�ȵ�����ͼ���ޣ����Լ����ӦΪ100%��n��20����
�����������������Ũ��Ϊ�����Ũ�ȣ����Լ����Ӧ����95%��n��20����
��/ǿ����������������Ũ�ȳ��жȵ�ǿ���ԣ����Լ����Ϊ100%����Ctֵ��CV��5%��n��20����
2.6�����
ע�������˸����Լ��м�ⷶΧѡ����Ӧ��HSV�ͱ���м������֤������HSV-1��HSV-2ͨ�ü����ͼ����Լ���Ӧ�ֱ����HSV-1��HSV-2������ͼ����ȷ������֤��ʹ������ȷHSV���ͺ�Ũ��/�ζȵĶ���������/�ٴ���������������������������ͼ���������Һһ�µ����Ի��ʽ���ϵ��ϡ�ͣ�������ͼ���Ľ�������֤��Ӧ�ṩ������������ٴ��������������Դ���Ʊ��������ͱ�ȷ�ϼ���ֵȷ�ϵ�������Ϣ��
2.6.1�����ȷ��
ÿ���ͱ�ѡȡ����3�ֲ�ͬ������/�ٴ�����������ֱ����ϵ��ϡ�ͻ�ö��Ũ���ݶȣ�ÿ��Ũ���ݶ��ظ���ⲻ����3�Σ���¼ÿ��Ũ���ݶȵĶ��Լ�����Լ�Ctֵ����100%�ɼ�������Ũ��ˮƽ��ΪԤ�����ޡ��ڴ�Ũ�ȸ����Ʊ�����Ũ���ݶ���Ʒ��ÿ��Ũ�������ظ����20�Σ�������95%���Լ���ʵ����Ũ����Ϊ��ͼ���ޡ�Ҳ�ɲ����ʵ���ģ�ͣ���Probit�������ͷ�������������95%���Լ���ʵ����Ũ��ˮƽ��Ϊȷ���ļ���ޡ�
2.6.2�������֤
�ֱ�ѡ����2.6.1��ͬ��Դ��HSV-1��HSV-2���������ٸ�3���ڼ����Ũ��ˮƽ������֤�������ٴ����Ի���ϡ�͵���ͼ����Ũ��ˮƽ�ظ�20�μ�⣬���Լ����Ӧ������95%��
ע��������Ӧ�ܹ��ṩ������ͼ������֤�IJ��������Դ��Ũ�ȼ��ͱ�ȷ���������Ϣ��Ӧ�Բ�ͬ�������ͼ���ͬ��������Һ�����漰���ֱ�������ͼ���ޡ�
2.7������
2.7.1����������Ϣѧ�����Բ�Ʒ���İ����Խ��з�����Ӧ�����ѹ����ĵ���������������С�
2.7.2�����Լ��м�ⷶΧѡ�����ʱ������������ԵIJ�ͬ��Դ��HSV-1��/��HSV-2�����ٴ���������ͼ�Ũ����ȷ��HSV-1��HSV-2�������������5�����а������о����о�����Ӧ�����ظ��ԡ�����ȡ�ע��������Ӧ�ܹ��ṩ���ڰ�������֤��������Դ���������͡��ͱ���ȷ��������Ũ�Ȼ��ζ�����ȷ��������������������ϸ��Ϣ�����ڼ����Ũ�ȸ�������δ�ܼ����������Ӧ�����Ũ�Ƚ��з�����ȷ������ͼ���ޡ���������֤�����ٴ�������������ͼ�����о������������ظ�ʹ�á�
2.8����������
2.8.1���淴Ӧ
ע������������Ӧ��Ա������������˻����鼰���ܴ�������ֳ��Ƥ��ճĤ������������ֳ���������������л������бȶԣ����ύ�ȶԽ��������ͬԴ��������Ӧ���н��淴Ӧ��֤��
����ʹ����Ӧ��ԭ�����Ե��ٴ������������˲�ԭ��������������ٴ��������н��淴Ӧ�о�����������Ӧ��Ԥ�ڼ����������һ�£�Ӧ��ÿ�����õ��������ͽ��н��淴Ӧ�о���
���淴Ӧ��֤��Ҫ���������Ŀ����ԣ��˻�����DNA��������λ���ܴ��ڵĶ�ֲ�����ɾ��Դ�����������ԭ�塢��������ͬ�������ٴ�֢״��������ԭ�塢�������о���ͬԴ�ԵIJ�ԭ�弰��Ҫ��֤������������ԭ��ȣ��Լ��ٴ�����м�����ϵ��������Ρ�
����֤������Ũ�����������DNA�������������Ϳ��ܴ��ڵ��������Ľ��淴Ӧ�����������������н��淴Ӧ�о�����*����ʾ������Ŀ, ��/����ʾѡ������һ��,�����С�������ʾ��Ӧ�ͱ����Ҫ��֤����
�����ࣺ������Σ������ͷ��������16��18��52��58)*�����������1��*�����������2��*��ˮ��-��״�����*��EB����*����ϸ������*����Ⱦ�����ಡ��*����������ȱ�ݲ���*������������70��71��*���ٲ�����3��7�ͣ�*���������6�͡����������7�͡����������8��*�������没���������ײ��������������˫���ɲ�����
ϸ���ࣺ�ܲ���ɪ��*��BȺ�����*���ſ�����Ѫ�˾�*����ѿ��Ĥ�˾�*�������ӵ��ɾ�*��������*�����������/���������������������˾�/������˾�/���϶���˾�/����İ����/�����־���/��ë����*����ϣ��*��������˾�*��������˾�*����Ƥ�������/�����������*���೦���*��ʺ�����*�����ɫ�������*��������״�˾�����С��״�˾������������˾����ܹ���֦�˾���������˾����������˾��������������
����ࣺ��ɫ�����*���⻬�����*��
����������֧ԭ��*����ֳ֧ԭ��*��������ԭ��*��ɳ����ԭ��ɳ���������*��ɳ����ԭ���Բ��ܰ���ѿ�ױ���*��÷�������������*������ë�γ�*���յع��γ档
�����ڲ�����ϸ����Ⱦ��ҽѧ���ˮƽ���н��淴Ӧ����֤��ͨ��ϸ����Ⱦ��ˮƽΪ106 CFU/mL����ߣ�����Ϊ105 PFU/mL����ߣ�Ҳ�ɲ�����������������ֵ��Ũ�ȣ��������Ũ��107 copies/mL��
ע��������Ӧ�ṩ�������ڽ��淴Ӧ��֤�������Դ����ɡ�����/�ͱ��Ũ��ȷ�ϵ��������ϡ�
2.8.2�������
ע��������Ӧ��ֿ����ٴ���������HSV1��HSV2�ϲ���Ⱦ�IJ�ԭ�壬�ڸ�Ũ�ȵ�����¶Ե�Ũ�ȣ����������Ũ�ȣ�HSV1��HSV2�������Ӱ�졣����HSV-1/2��HSV-2/1�ĸ��ţ�HIV��HPV��HSV-1��/��HSV-2�ĸ��ż��������ܹ���Ⱦ�IJ�ԭ���HSV�ĸ��ţ�������ע�������˽���걨�Լ��ķ�Ӧģʽ�����ÿ��ܻ�ϸ�Ⱦ�ĸ�Ũ�Ȳ�ԭ�����ͼ����Ũ�ȸ�����HSV-1��HSV-2�ֱ������������о���
2.8.3�����Ը���
�����Ը����о��IJ�ԭ����Ͻ���Ϊͬһ��Ӧ��ϵ�ڲ�ԭ�塢������֢��Ⱦ��ԭ�弰������ϸ�Ⱦ��ԭ�塣����HSV1��HSV2�Ķ��غ����⣬ע��������Ӧ��֤HSV-1��HSV-2�֮��ľ����Ը��š�
2.8.4������������
Ӧ��Բ�ͬ�������ͣ��ֱ����ۿ��ܴ��ڵĸ��������������õ���������ٽ�����������������������ÿ�ָ������ʵ�DZ�����Ũ�ȣ�������������������½��и������顣HSV-1��HSV-2��ֱ���и������о����ṩ�������о������鷽�����������������ŵĽ��ܱ����Խ�����к�����ͳ�Ʒ������Ա����Ӹ�������ǰ���Ctֵ���졣DZ�ڵĸ������ʰ�����Դ�ԡ���Դ�ԡ��������ɼ��ʹ����ڼ���������ʼ������ѱ����ĸ������ʡ�
������ֳ����������Ӧ����ѡȡȫѪ����Ѫ�쵰�Ͱ�ϸ�������˰��ס������ء�����ճҺ�Ƚ����о���
����ҩ��ĸ��ţ��Ļ��ء�����ù�ء�����ù�ء���ù�ء���ŵ���ء��ǰ���ड��⻯�ɵ��ɡ��������ɡ�������Τ��������Τ������ת¼����ҩ��ȿ�����ҩ���������ҩ������ҩ������ҩ��������������ҩ�������ҩ���Ů��������Ʒ������˨������������������ϴ���Լ�������������λҩ��ij���ҩ�
�����������͵ĸ������о�Ӧ���������������Ϳ��ܺ��еĸ������ʽ�����Ӧ�о������ṩ�����PŨ��ѡ�����ݡ�ע��������Ӧ�Բ��õIJ�ͬ������ȡ�Լ��������ɼ����������������ӡ������ܡ���������Һ�ȣ������������������̿�������ĶԼ��ϵͳ�ĸ������ʽ��г�ֵĸ������о���
2.9 HSV����DNA��ȡ
�ڽ��к�����֮ǰ�������к��ᣨDNA����ȡ/�������衣�ò����Ŀ�ij�����������Ŀ��DNA�⣬��Ӧ����Ӧ�Ĵ������ã�������ȥ��PCR����������걨��Ʒ�Ƿ���DNA����/��������֣�ע�������˶�Ӧ�����ʹ�õ����к�����ȡ�Լ�������ȡ���ᴿ�ȡ�Ũ�ȡ���ȡЧ�ʵ��о������������Ϻõĺ�����ȡ�Լ�����ƽ�бȶԡ�����Ʒ�������ֻ����Ϻ�����ȡ�Լ�����ÿһ�ֺ�����ȡ�Լ�������ϼ���Լ����п����š����ܶȺͼ������֤�������������ȡ�Լ���ȡԭ�����ڲ��죬���������м���Ľ����о���
2.10��Ӧ��ϵ
2.10.1�����ɼ��ʹ���
2.10.1.1���������ɼ���λ���ɼ���ʽ������������ʽ��ѡ���ṩ��ص��о����ϡ��������������ռ����Ʊ����������䣨���漰���ʹ���ķ�ʽ������֤����Ҫʱ����ѡ���ȷ�ϲ��ṩ��ص��о����ϡ���ͬ���͵�����Ӧ�ֱ�����о���
2.10.1.2�����������Ӽ���������Һ��ѡ����������ɼ����ӵ���������֤�����Ӷ�������ϴ��Ч�ʡ��ṩ��ϸ�ı���Һ���ѽ�Һ�ijɷ֡�Ũ�ȡ�ʹ������Ҫ����������֤���ϵȡ����IJ�ͬ����Һ���ѽ�Һ����֤������ظ��ԡ�
2.10.2������ȡ�ͷ�Ӧ��ϵ
�о�ȷ����Ѻ�����ȡ�ͷ�Ӧ��ϵ������������ȡ�õ����������ϴ�������PCR��Ӧ�������������øŨ�ȡ�����/̽��Ũ�ȡ�dNTPsŨ�ȡ�dUTPŨ�ȣ����У���������Ũ�ȼ���Ӧ�����¶ȡ�ʱ�䡢ѭ������ӫ��ɼ���������Ӧ����ȡ������ڱ�֤������ȡ����������¾��������ܷ�Ӧ��ϵ�ͼ������������������ȡ�
�ύ��ͬ���û��ͻ��ߺ���ֵѭ������ȷ�����ϡ�
��ͬ���û��͵ķ�Ӧ��������в���Ӧ�ֱ����������ύ��֤���ϡ�
2.11�ύЯ����Ⱦ�о����ϣ������ã������ú����ļ��˳��Զ����Ũ������������������������������Я����Ⱦ��
3.�ȶ����о�����
�걨�Լ����ȶ�����Ҫ����ʵʱ�ȶ��ԡ������ȶ��ԡ���ƿ�ȶ��ԡ������ȶ��ԣ������ã������ڴ������Ƶ��о���ע�������˿ɸ���ʵ����Ҫѡ��������ȶ����о��������ȶ����о�����Ӧ�����о�������ȷ�����ݡ������ʵʩ��������ϸ���о������Լ����ۡ�����ʵʱ�ȶ����о���Ӧ�ṩ����������Ʒ��ʵ�ʴ��������±�������Ʒ��Ч�ں���о����ϡ����ڿ�ƿ�ȶ����о�Ӧģ����ʵʹ�����Σ�������ƿ�ȶ��ԵĿ�ƿƵ�κͿ�ƿʱ��ȡ�
4.�����ж�ֵ�о�����
�����ж�ֵȷ��������Ҫָ���걨��Ʒ���ڽ���жϵĺ������Ctֵ��/�����Ũ���ٽ�ֵ����ȷ�ϵ����ϡ�����ȷ�����ߡ���ֵ����ֵѭ������Ct��������Ũ�ȣ������ã��ȵ��о����ϵȡ����ж�ֵ���ڻ�����Ӧ�ṩ������ȷ�����ϡ�
�����ж�ֵ�о�������ԴӦΪԤ����Ⱥ�������Dz�ͬ�Ա����䡢������������أ������ܿ���������Դ�Ķ����ԡ������ԣ������걨��Ʒ���ǵ��������͡�Ƥ��λ�������ã�����������϶������Ե���������HSV-1��HSV-2Ӧ�ֱ����ͳ�Ʒ��������ڽ��걨���һ�����͵��Լ���ע��������Ӧ������һ��������Ϊ������������ͳ�Ʒ������ύ�����ж�ֵ�о����������ı�����Ϣ�б������ٰ����Ա����䡢�������͡�Ƥ��λ�������ã����ٴ������Ϣ��������Դ���������������Ϣ������������о�������Ӧ˵��������ԡ�
���⣬����ע�������˿��ǽ��������ж�ֵʱʹ�õ���������������Ŀ����Ⱥ�Ĵ����ԣ���ͨ���ٴ����۽�һ����֤��ȷ�������ж�ֵ��ȷ�ԡ�
�����Ʒ���ò�ͬ�������ͣ�Ӧ�Ը��������ͽ��������ж�ֵ����֤��
�ṩ�ڱ�������Χ��ȷ���������о����ϡ�
5.��������
5.1��Ҫԭ�����о�����
�����Ʒ����Ҫԭ���ϰ������̽�롢ø��dNTP���������/������֣����У�����ҵ�ο�Ʒ������Ʒ���ʿ�Ʒ�ȡ�Ӧ�ṩ��Ҫԭ���ϵ���Դ��ѡ���Ʊ��������о����ϣ���������֤�飬��Ҫԭ�������������ƶ��ͼ������ϡ�����Ҫԭ����Ϊ��ҵ���ƣ�Ӧ�ṩ����ϸ���Ʊ����������������ƹ��̣����Ʊ����ձ�������ȶ�������Ҫԭ����Դ�����Ӧ�ṩ���ϰ�����ѡ���ԭ���ϵ����ݼ��Ա�ɸѡ�������ϡ�ѡ���Ĺ�Ӧ�����ƣ���Ӧ���ṩ����������ԭ���ϼ��鱨�棨����֤�飩���Լ���ԭ���ϵ�������볧���鱨�棬��Ӧ��ӦΪԭ���ϵ������̣��������������ע��������Ӧ�Ը���Ҫԭ���Ͼ���ȷ�������Ʊ���
5.1.1�������/������֣����У�����Ҫ��ɡ�ԭ�����ܼ���ص���֤���ϡ�
5.1.2�����̽��
Ӧ���������̽������ԭ���������Ժ������ԵĿ���������ṩ���̽��������С�ģ��������м����ߵĶ�Ӧ���������ÿ�ֲ����������������̽���Թ�ɸѡ���������Ԥ�����õĻ����ͱ���м�������������ԣ��罻�淴Ӧ�������ۣ�ѡ�������ϣ����ύɸѡ���о����ݡ����̽���������Ӧ���ٰ�������ȷ�ԡ����ȡ�Ũ�ȡ�̽���ǵ�ӫ���ؼ�����������ȡ�ע��������Ӧ�ṩ��֤���ϻ�ϳɻ������ߵ��ʼ�֤������PAGE��Ӿ������ЧҺ��ɫ����HPLC������ͼ�ȡ�
5.1.3������������գ�dNTPs��
������dATP��dUTP��dGTP��dCTP��dTTP��Ӧ�ύ���䴿�ȡ�Ũ�ȡ������ȶ��Եȵ���֤���ϡ�
5.1.4ø
��Ҫ��ø��Ҫ����DNA�ۺ�ø�������DNA�ǻ���ø�����У��ȣ�Ӧ�ֱ��ø���ԡ������ԵȽ������ۺ���֤��
DNA�ۺ�ø��Ӧ����DNA�ۺ�ø���ԣ���������ø���ԣ������ȶ��ԣ��磺94�汣��1Сʱ���Ա���50%���ԡ�����ø��������ȷ������ø���ԡ�
�����DNA�ǻ���ø��UDG/UNG��������ˮ����������ռ��Ļ��ԣ���������ø����������ø���ԡ�Ӧ��ø���Լ����ȶ��Խ��к�����֤��
5.1.5���������Լ��İ�װ���ϺͺIJ�Ӧ���������Ǻ���ø��DNase���ͺ��Ǻ���ø��RNase����Ⱦ��
5.1.6��ҵ�ο�Ʒ��
�����ã�Ӧ�ύ��ҵ�ο�Ʒ��ԭ����Դ����ɡ�ѡ���Ʊ�����������(�����ơ��ͱ�)����/���Լ�Ũ��/�ζ�ȷ�Ϸ������Լ��������֤���ϣ���Դ����ֵ���ϵ���Ϣ������ҵ�ο�Ʒ�ĺ�������Ӧ���ƷԤ�ڼ��İ�����һ�¡���ҵ�ο�Ʒ�Ļ������ٴ������ռ�/�������һ�¡�������ò����ꡢ�ٴ�����������ٴ����������ο�Ʒ������ʹ���������ٲ�������������ȡ/������ȡ�
��ҵ�ο�Ʒ����ĿӦ���������Բο�Ʒ�����Բο�Ʒ����ͼ���ο�Ʒ�����ܶȲο�Ʒ�ȡ�
���Բο�Ʒ������Ӧ���ؿ��Ǽ����������֤��������ö����ͬ������Դ��HSV�������������Ʊ��������ò�ͬ�ζ�ˮƽ�������Լ����ܷ����HSV���ͣ����Բο�Ʒ��Ӧ����������õ�HSV���ͷֱ����á�
���Բο�ƷӦ���Ƿ��������Ե����ۣ�Ӧ���������ٴ����������������ص������������Լ��м�ⷶΧ�ڵ�HSV��������������������֢״�ġ�����Ⱦ��������ԭ��������Ʒ��
��ͼ���ο�Ʒ��ע��������Ӧ��ȷ����ο�Ʒ�в�������Ũ�ȵ�ȷ����������ȷ����ο�Ʒ�в�������Ũ�ȵ�ȷ�����ݣ�����ο�Ʒ��HSV����Ũ��ӦΪ�걨��Ʒ�����Ũ�Ȼ��Ը��ڼ����Ũ�ȡ�
���ܶȲο�ƷӦ���ٰ�������Ũ��ˮƽ�����а���������ˮƽ��
5.1.7�ڶ��գ��ڱ꣩���ʿ�Ʒ
�ڶ��գ��ڱ꣩����Ӧ��������/�����ʿ�Ʒ�˲��û���ٴ����������ٲ����Ʊ���
�ڱ��ԶԹ������ƿ�����ɵļ����Խ�������ʿء�ע��������Ӧ���ڱ�����̽���ģ���Ũ������ȷ��֤����֤�ڱ�ӫ��ͨ�������Ե��������ߣ���Ӧ��Ŀ�Ļ���ļ����ɾ��������ƶ����¼����ԣ����ڱ��CtӦ����ȷ�ķ�ΧҪ��
��/�����ʿ�ƷӦ�������������ƽ����ȡ���Զ�����PCR��Ӧ���̡��Լ�/�豸��������Ⱦ�Ȼ��ڽ��к����������ơ�ע��������Ӧ�Ը����ʿ�Ʒ��Ct������ȷ�ķ�ΧҪ��
5.2�������������
���ܲ�Ʒ��Ҫ�������գ���ȷ�ؼ��������Ƶ㣬��������ͼ������ֵķ�ʽ�������ύ��Ҫ�������յ�ȷ�����Ż��о����ϡ�
���ģ��ٴ�����
�ٴ�����Ŀ�չ���������ƶ��Լ������д�Ⱦ�Ӧ������ط��漰����������Լ��ٴ����鼼��ָ��ԭ��Ҫ�������˵�������Ʒ�ٴ�������Ӧ��ע���ص����⡣
1.�����
��������ͬ���Ʒ���е��Լ����ٴ��о���ѡ�����������С��ٴ��ձ���Ϊ�����Ϻõ�ͬ���Ʒ��Ϊ�Ա��Լ�������������������Լ���֮���жԱ������о������۱���Ʒ���ٴ����ܡ�
2.������Ⱥ
�ٴ�����������Ӧ�������ֿ����е����������Ⱦ����Ⱥ�����е����������Ⱦ����֢״�Ļ��ߣ�����ֳ������������ʹ������ȡ����Ժ�Ů��������Ӧ�������ȷֲ���
3.�ٴ����鲡����
�ʵ����������DZ�֤������������Լ��ٴ����ܵõ�ȷ���۵ı�Ҫ�������ٴ�����������Ӧ����ͳ��ѧҪ�ɲ����ʵ���ͳ��ѧ�������й��㡣������Ӧ�ٴ�������ƣ���ѡ�����Է�����/����Ⱥ����Է�����/�����ȣ��ֱ�����������������������������������
����������ֳ�����ӣ���Ů�Թ������ӻ��������ӡ�����������ӣ������ٴ��������������������Լ���������ͬ���Ʒ���бȶԵ�������ƣ��ɲ��õ���Ŀ��ֵ����������ʽ���������������
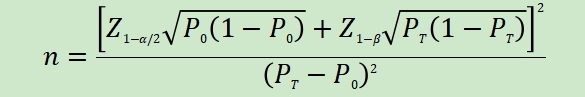
��ʽ�У�nΪ��������Z1-��/2��Z1-��Ϊ������ˮƽ�Ͱ��նȵı���̬�ֲ��ķ�λ����P0Ϊ����ָ����ٴ��ɽ��ܱ���PTΪ������������Լ�����ָ��Ԥ��ֵ��
����������֪������ָ����ٴ��ɽ��ܱ���P0����HSV-1/HSV-2�����Լ����Է����ʺ����Է�����ԭ���Ͻ��鲻����95%��������ָ��P�ӽ�100%ʱ���������������㷽�����ܲ����ã�Ӧ����ѡ��������˵ķ����������������㣬�羫ȷ���ʷ��ȡ����ٴ������о��и����������������㷽ʽ����˵��������Ժ���ɲ��á�
�����������������Ҫ�����ٴ������в������������ʣ�һ����ԣ������������ʲ�Ӧ����10%�����⣬�ٴ�����Ӧ����һ��������������������
������ֳ������Ƥ���ĤƤ��걾������������Һ���ӣ���Һ�����ٴ�����������Ӧ�����ʵ���ͳ��ѧ�������й��㣬����ϸ������ʹ��ͳ�Ʒ���������������ȷ�����ݡ�
���걨��Ʒ���ж������õ��������ͣ���ͬ��������֮����ڽϴ���죬���磺�������ܲ�ͬ���ٴ����ܲ�ͬ��������Ⱥ��ͬ����Ӧ֤��ͬ�������ж�ֵ��ͬ�ȣ��������Ҫ�ֱ�����ٴ�������ƣ������ֱ���������������ͳ��ѧ������
�������Ʒ����������/��Ĥ�����������Ӧ֤��Ӧ���ݲ�ƷԤ����;���ۺϿ��Dz�Ʒ���ս��п�ѧ���ٴ�������ơ�
4.�ٴ��о���λ��ѡ��
Ӧѡ������3�ң���3�ң��ٴ����������������ط��桢ָ��ԭ���Ҫ��չ�ٴ����顣�ٴ����������ѡ��Ӧ��������������������Լ����ص��Ԥ����;���ۺ����в�ѧ�����������ߵ�ѡ�����һ���ĵ�������ԡ����ٴ��������Ӧ���з�������ѧ�����������ƣ�����������ԱӦ���㹻��ʱ����Ϥ���ϵͳ�ĸ����ڣ���Ϥ���۷�����
5.�ٴ���������ͳ�Ʒ���
Ӧѡ����ʵ�ͳ�Ʒ������ٴ�����������ͳ�Ʒ���������������������Լ���Ա��Լ�/������һ�������ۣ�һ��ѡ��2��2������ʽ�ܽ������Լ�/�����ļ���������ݴ˼������Է�����/�����ȡ����Է�����/����ȡ��ܷ����ʡ�Kappaֵ��ָ�꼰���������䡣
���ڲ�һ��������Ӧ����ԭ����������ٴ����鷽���涨����������������ȷ�ϣ���ȷ�Ͻ����Ӧ����ͳ�Ʒ�����
6.����ѧҪ��
�ٴ����������Ϻն��������Ե�����ѧ�������ٴ������������ίԱ���ͬ�⡣�о���Ӧ��ֿ����ٴ������������Ļ�ú��������������ߵķ����ԣ�Ӧ�ύ����ίԱ�����������
7.��������
�ٴ����鿪ʼǰ����������ٴ��������ѵ������Ϥ������������鷽���IJ������������������ܵȣ�����ȿ�������������������̶�Ӧ������Ч�����������£�����ȱ�֤�������ݵ�ȷ�Լ����ظ��ԡ�
8.�ٴ����鷽��
��������Լ��ٴ�����Ӧ����ͬһ�ٴ����鷽���ڶ���ٴ����������չ���ұ�֤�������ٴ������������ѭԤ���ķ�������������Ķ��������������Ӧ���ٴ����������ʵ�����ڲ��ɸ�ʵ���ҵļ�����Ա������ɣ��걨��λ�ļ�����Ա�����б�Ҫ�ļ���ָ���⣬�����������ʵ����̡�
���鷽��Ӧȷ���ϸ����ѡ/�ų������κ�����ѡ���������ų����ٴ����鶼Ӧ��¼�ڰ�����ȷ˵��ԭ��������������̺ͽ���ж�ʱ��Ӧ����ä���Ա�֤�������Ŀ��ԡ����ٴ��������ѡ�õĶԱ��Լ�/����Ӧ����һ�£��Ա���к�����ͳ��ѧ���������⣬Ӧ����������������Լ�����������Ա��Լ�/�����������͵Ŀɱ��ԣ����в��죬���ṩ��ֵ���֤��
9.�ٴ����鱨��
Ӧ�������������Ƽ������ؼ�����������������IJ�����Ӧ�ö������ٴ�����ʵʩ���̡�������������۵Ƚ���������������������Ӧ������Ҫ�����ݺ�ͳ�Ʒ���������
���壩��Ʒ˵����ͱ�ǩ����
��Ʒ˵�����ʽӦ���ϡ���������Լ�˵�����дָ��ԭ��Ҫ��Ʒ˵�����м������ݾ�Ӧ��ע���걨�����е�����о��������һ�£���ijЩ���������Բο����ף�Ӧ�Թ淶��ʽ���б�ע���������������������Ϣ��
HSV���������⼰�����Լ�˵�����дӦ�ص��ע�������ݡ�
1.��Ԥ����;��
1.1�Լ����������ⶨ�Լ��XX�����е�HSV-1��HSV-2 ���ᣨDNA������ɷ���������ȷ��������������Ӧ�����걨��Ʒ�ķ��������������ٴ��о��������ȷ�ϡ�
1.2��Ʒ���ܣ����Ŀ����Ⱥ���ٴ����ֺ��������ָ�꣬��������ֳ�������е����������Ⱦ�ĸ�����ϡ�
1.3 �ٴ�������Ϣ����Ҫ������ԭ������ѧ�������²��ԣ����в�ѧ��������Ⱦ���ٴ����ּ���ؼ����ȡ���Ҫ����HSV��ص��ٴ���ʵ������Ϸ�����
2.������ԭ����
����������ȡ�������Լ��м��ԭ������ȷ��Ʒ�ܹ�����Ŀ�������Ϣ��������������ȷ�ڱ�������Ƽ������ã���ȷ����̽����Ƽ�̽����ӫ����Ϣ������˷���Ⱦ��ʩ�����м�Ҫ������
3.����Ҫ��ɳɷ֡�
��ȷ�Լ����и���ּ�����ɷ֣���ȷ��Ҫ��δ�ṩ�IJ��ϣ��������ȡ�Լ��������ɼ��ܡ����ӡ���������Һ����������ʹ�õIJ��ϵIJ�Ʒ���ơ��������ҡ����ż�ע��֤�Ż��ŵ���Ϣ��
4.������Ҫ��
4.1�����ɼ�Ҫ���������ɼ�ʱ����ѡ���Ƿ����ٴ�֢״����ҩ��������ص�Ӱ��ȣ�
4.2�����ɼ���������Բ�ͬ���������ͷֱ���ϸ���������ɼ��������豸Ҫ�����������衢�Բ������ӡ��������������ͱ���Һ��Ҫ����������Һ���������Ҫ��ȡ�������ȷ����֤������ʹ�õ����������������������Ӻͱ���Һ����Ϣ�������ٴ������Ƽ��IJ���Ҫ��Ӧ��ѭ����������Ӧ�ļ����淶��ָ�ϡ�
4.3�������������ͼ����棺�����о����ϣ���ȷ������������ȡǰ��Ԥ��������������������������������Ϣ���������õ����Σ���Բ�������뱣��Һǰ�ĸ������ӡ�������������Һ�������ȡǰ��������ȡ�������Ȳ�ͬ�εĴ�����ʽ�������ȶ��Լ����������ȡ��������������䶳������ȷ���ڴ�����
5.�����鷽����
��ϸ˵����������ĸ������衣
��ȷ������ȡ�õ����������ϴ�������PCR����������������ʿ�Ʒ���������ͬ�����к�����ȡ��������ȷ�����û��͵ķ�Ӧ�������ã����ߡ�ѭ����ֵ��Ctֵ����ѡ�����Լ������б��Ӧ��ӫ��ͨ������ȷ�ڱꡢ�ʿصļ����Ctֵ��Χ��
6.���������Ľ��͡�
������Զ��ա����Զ��ա��ڶ��գ��ڱ꣩�Լ�Ŀ�����ļ������ͨ���������ߺ�Ctֵ���������Ե��жϣ��������п��ܳ��ֵĽ����ϼ���Ӧ�Ľ���Ct������ڻ�����Ӧͬʱ˵���Ի����Ĵ�����ʽ�������ں����������Ҫ�����ظ���⣬�ظ����ķ��������������ܲ�ȡ���Ż���������ɼ�Ҫ���ɼ��������ȡ�
7.�����鷽�������ԡ�
7.1������������ٴ��ο�������ȷ�ﲡ�������ٴ�֢״����������ֶΡ�
���Լ��еļ����Ӧ��ϻ��ߵ�֢״/��������ʷ������ʵ������Ͻ������������ۺϷ����Լ����ͣ�������Ϊ�����ٴ����λ������Ψһ���ݡ�
7.2 HSV DNA������Բ����ų�HSV��Ⱦ��
7.3���¼�����/�����Խ���Ŀ����Է�����
7.3.1�������������ɼ������������估����������������Ŀ����ζȹ��͡�
7.3.2 HSV��������Ŀ��������еı��������ԭ���µ����иı䡣
7.3.3ͬһ���߲�ͬʱ�䡢��ͬ��λ���߶�βɼ������ή�ͼ����Խ���Ŀ����ԡ�
7.3.4δ����֤���������ţ�����Դ�Ի���Դ�������������ʡ�
7.3.5������Ľ�����Ⱦ��
7.3.6δ����֤���������淴Ӧ���ʡ�
8.����Ʒ����ָ�������������ָ�꣺
������Ʒ���ܣ������������ݣ�
8.1 ���ұ�Ʒ�����У�����ҵ�ο�Ʒ�ķ����ʡ�
8.2 ����ޣ�˵����ͬ������������ͼ���ޣ���Ҫ������ͼ����ȷ�������Լ���֤��ͼ���������õľ�����͡���HSV-1��HSV-2��ͼ����ͬ��Ӧ�ֱ��г���
8.3���ܶȣ�˵����ͬ�����������ظ��Ժ����������۽����
8.4���������ԣ��������淴Ӧ�������ʡ�
8.4.1���ܲ������淴Ӧ��������ԭ�����֤������������б��ķ�ʽ������ԭ�����ơ��ͱ�Ũ�ȵ���Ϣ��
8.4.2�����г����������ʶԼ������Ӱ�죬Ӧע���ɽ��ܵ������ֵ��
8.5�ٴ����飺��Ҫ�������鷽�����������������������ͽ��۵ȡ�
9.��ע�����Ӧ���ٰ����������ݣ�
9.1���Ʒ������Դ����Դ�����ʣ�Ӧ�ṩ����DZ�ڴ�Ⱦ�Եľ�ʾ����Ϣ��
9.2�ٴ�ʵ����Ӧ�ϸ����ء�ҽ�ƻ����ٴ�������������ʵ���ҹ����취�����ٴ���������ʵ���ҵĹ����淶����������ʵ��Ӧ�ɾ���רҵ��ѵ��ʵ������Ա�ϸ���˵����Ҫ����С�
9.3ǿ����Ʒ���ܽ�������Ƶ������������ͼ�������Ҫ������˵���������ɼ��ʹ��������������������ӡ������ɼ�Һ�ȣ���������֤�������������ͻ������ɼ��������������ܱ�֤��Ʒ���ܡ�
���������
[1]�����г��ල�����ܾ�.��������Լ�ע���뱸�������취: �����г��ල�����ܾ����48��[Z].
[2]����ҩƷ�ල������.���ڹ�����������Լ�ע���걨����Ҫ�����֤���ļ���ʽ�Ĺ���:����ҩƷ�ල������2021���122��[Z].
[3] ����ҩ��� ������������ί. ҽ����е�ٴ��������������淶: ����ҩ��� ������������ί2022���28��[Z].
[4]����ҩƷ�ල������.���ڷ�����ҽ����еע���Լ�����涨���Ĺ���:����ҩƷ�ල������2021���126��[Z].
[5]����ҩƷ�ල������. ��������Լ��������:����ҩƷ�ල������2021���129��[Z].
[6]����ʳƷҩƷ�ල�����ܾ�.��������Լ�˵�����дָ��ԭ��.����ʳƷҩƷ�ල�����ֹܾ���2014���17��[Z].
[7]����ҩƷ�ල������.��������Լ��ٴ����鼼��ָ��ԭ����ҩƷ�ල������2021���72��[Z].
[8]����ҩƷ�ල������.ҽ����е��Ʒ����Ҫ���дָ��ԭ����ҩƷ�ල������2022���8��[Z].
[9]����ҩƷ�ල������ҽ����е������������.���Լ����������Լ�������������ע�����ָ��ԭ����ҩƷ�ල������ҽ����е������������2022���36��[Z].
[10]WS/T 236-2017����ֳ���������[S].
[11]���������ʵʱӫ��PCR����������2�棬��ѧ�����磬2016[M].
[12]WHO. Laboratory diagnosis of sexually transmitted infections, including human immunodeficiency virus.2013 [Z].
[13]WHO. Guidelines for the treatment of Genital Herpes Simplex Virus. 2021[Z].
[14]WHO. Guidelines for The Management of Symptomatic Sexually Transmitted Infections, 2021 [Z].
[15]CDC. Sexually Transmitted Infections Treatment Guidelines, 2021 [Z].