

| ��ָ��ԭ��ּ��ָ��ע�������˶������ϸ����ԭB27��HLA-B27���������Լ�ע���걨���ϵ�����д��ͬʱҲΪ�������������ṩ�ο�����ָ��ԭ���Ƕ�HLA-B27�������Լ���һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ������� |
��ָ��ԭ��ּ��ָ��ע�������˶������ϸ����ԭB27��HLA-B27����������Լ�ע���걨���ϵ�����д��ͬʱҲΪ�������������ṩ�ο���
��ָ��ԭ���Ƕ�HLA-B27��������Լ���һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����á��������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ����
��ָ��ԭ��Ϊע�������˺ͼ���������Աʹ�õ�ָ�����ļ����������������������漰������������Ϊ����ǿ��ִ�У�Ӧ����ѭ��ط����ǰ����ʹ�ñ�ָ��ԭ��������ܹ�������ط���Ҫ�������������Ҳ���Բ��ã�������Ҫ�ṩ��ϸ���о����Ϻ���֤���ϡ�
��ָ��ԭ���������з���ͱ���ϵ�Լ���ǰ��֪ˮƽ���ƶ������ŷ���ͱ��IJ������ƣ��Լ���ѧ�����IJ��Ϸ�չ���������Ҳ����ʱ���е�����
һ�����÷�Χ
ǿֱ�Լ�������һ���������Լ�����������������׳�����ԣ���Ҫ�ַ����Ĺؽڡ�������ͻ������������֯�����ܹؽڣ��ɰ鷢ǰ����Ĥ�ȹؽ�����֣������߷����������κ�ǿֱ��
HLA-B27����λ�������6��Ⱦɫ��̱�HLA����B�������и߶Ȼ����̬�ԣ���������ΪHLA-B27��ԭ��HLA-B27 ��ԭ������ǿֱ�Լ����߶���أ�����90%��ǿֱ�Լ������ߵ�HLA⁃B27 ��ԭ����Ϊ���ԣ�����ͨ��Ⱥ�н�5% ~10% Ϊ���ԡ�HLA-B27�����������ؽ�������Ҳ�в�ͬ�ļ�����ڷ�Ӧ�Թؽ����������ʿɴ�60%-90%������м���ؽ������Գ����ؽ����������ʾ��ɴ�50%-60%��
HLA-B27Լ��167���������ͣ������ͷֲ����ڵ�������ֲ��졣HLA-B*2705��ȫ��ֲ��������ͣ�����Ϊ�������������͵���������й�������Ⱥ�У�HLA-B*2704��HLA-B*2705����Ҫ�������ͣ�ͬʱ������HLA-B*2702��HLA-B*2703��HLA-B*2706��HLA-B*2707��HLA-B*2709��HLA-B*2702��HLA-B*2704��HLA-B*2705��HLA-B*2707�Ǽ����л���HLA-B*2703��HLA-B*2706��HLA-B*2709�뼲���Ĺ�ϵ�д������顣
��ָ��ԭ�������ڻ���ӫ��PCR�������Լ���˾���ȫѪ�����е�HLA-B27������Լ�������ǿֱ�Լ����ĸ�����ϡ�
���ڲ���������������ѧ��ⷽ�������������������������͵ļ���Լ������ܲ���Ҫ����ȫ���û����������ݲ���ȫ�棻�����˿ɲο���ָ��ԭ��ͬʱ���ݲ�Ʒ���Զ����ò��ֽ������ۣ��Բ����ò��ֲ��������õ����ɣ�����֤��������Ŀ�ѧ�����ԡ�
��ָ��ԭ�������ز�Ʒע���걨�����еIJ������ݽ���д������δ������Ӧ��������ط���Ҫ��
����ע�����Ҫ��
��һ�������Ϣ
1.��Ʒ���Ƽ��������
��Ʒ����Ӧ���ϡ���������Լ�ע���뱸�������취������ط����Ҫ����ͬʱ���������˲ο�������ͬ���Ʒ�IJ�Ʒ���������ݡ���������Լ���������ò�Ʒ���յ�������������Լ��������������Ϊ6840��
2.������Ϣ��������Ʒ�б���Ӧ����ҽ����еΨһ��ʶ���������ļ����걨ǰ���ܻ�������ϵ�����ͨ��¼�Լ��������������ļ���
��������������
����������Ҫ������������Ʒ������Ԥ����;���걨��Ʒ������ʷ��������˵�������ݡ�����,��Ʒ����Ӧ��������ԭ������Ʒ��Ҫ�о�������ܽ�����ۡ���ͬ���/��ǰ����Ʒ�ıȽϵȡ���ͬ���/��ǰ����Ʒ�ıȽ�Ӧ���ش�Ԥ����;������ԭ������Ҫ��ɳɷ֡�����ָ�ꡢ�ٴ�Ӧ������ȷ�����ϸ˵���걨��Ʒ���ѻ�����ͬ��/ǰ����Ʒ֮�����Ҫ����
���������ٴ�����
1.��Ʒ����Ҫ���鱨��
ע��������Ӧ����ԭ�������������������ȶ���ǰ���£����ݲ�Ʒ���ơ�ǰ�����۵Ƚ������������ļ����ϣ���ϲ�Ʒ�����ա�ҽ����е��Ʒ����Ҫ���дָ��ԭ��Ҫ���д�������Ʒ��Ϊ��������������Լ���Ӧ���Ը�¼��ʽ��ȷ��Ҫԭ�����Լ���������Ҫ��
��������������Լ�Ӧ���ṩ������ͬ�������β�Ʒ�ļ��鱨�档Ŀǰ�������õĹ��ұ�Ʒ����������Ҫ����Ӧ���ֹ��ұ�Ʒ�����Ҫ���ύ������һ��ʽ�ļ��鱨�棺
��1�������˳��ߵ��Լ챨�档
��2��ί�������ʵ�ҽ����е����������ߵļ��鱨�档
�������õĹ��ұ�����ҵ������Ʒ����Ҫ������Ҫ��Ӧ��������Ӧ��Ҫ��
2.���������о�
ע��������Ӧ�ύ�ڷ�������������ϵ�Ļ������������Լ��н��е����з��������������ϣ������������鷽�����������ݡ�ͳ�Ʒ�����������۵���ϸ���ϡ��й�����ı�����ϢҲӦ���걨�����н�����������������ص㣬���õ��Լ����ơ��������ţ��������ƺ��ͺţ������ı�����Ϣ����Դ��������š��������͡��ɼ���������ʽ�������ͺ�Ũ��ȷ�Ϸ�����������ȡ�������������������ӦΪ��ʵ�����������ƶ��������������ͣ�ע��������Ӧ��Բ�ͬ�������ͷֱ�������������о���
�����������������鷽�����Բο����ʻ�����й���������Լ�����������ָ��ԭ����У����ڱ����Ʒ�������ض����·������ܽ����о���
2.1�����ȶ��Լ����õ���������
Ӧ��ֿ��������ɼ�/���������䡢����ȸ����ڵ�Ӱ�죬ȷ�������ı�������������ʱ�䲢�������ȶ��Խ��г����֤����������ȡ���������м�⣬�������ȡ���DNA�ȶ��Խ��г���о��������ƶ��������������ͣ�ע��������Ӧ��Բ�ͬ�������ͷֱ���������ȶ����о���
����ȫѪ�����������ö��ֿ�����������ͨ��ͬԴ�ȶ��о���һ���������ٴ�������֤���ֿ������������ԡ�
2.2�ʿ�Ʒ�ĸ�ֵ
�����ʿ�Ʒ�ĸ�ֵ���̣����ύ������Ʒ�ڲ�ͬ���û����ϵĸ�ֵ�о����ϡ�
2.3ȷ��
��ͨ���걨��Ʒ���������������ͬ���Ʒ���з���ѧ�ȶ��о��������걨��Ʒ��ȷ�ȣ�Ҳ��ͨ�����ο�Ʒ���̣������걨��Ʒ������뾭ȷ�Ͻ���ķ�������������걨��Ʒ��ȷ�ȡ�
����ȷ�ȵ�����Ӧ�����ɷ��ٴ�������ɣ���������Ӧ���Ʒ���÷�Χһ����������ͬŨ��ˮƽ��
2.4���ܶ�
�����ظ��ԡ��м侫�ܶȺ������ԡ�
���ܶ�����Ӧ������ʵ���������о��������������������������������������ǿ���������������Ӧ����˵����ִ�У������������ȡ/�����������������衣
���ܶ���������������Ҫ��
2.4.1�Կ���Ӱ���⾫�ܶȵ���Ҫ���ؽ�����֤��������Լ�������������ȡ/������֣������⣬�����������������ߡ��ص㡢ʱ�䡢����ִΡ��Լ����εȡ�
2.4.2�趨�����ľ��ܶ��������ڣ���������/���䡢����/�ռ䡢��ͬ������Ͳ�ͬʵ���Ҽ�ľ��ܶȽ����ۺ����ۡ�
2.4.3�ɽ�ϲ�Ʒ���ص㣬�Ծ��ܶ�ָ�����۱���������Ҫ����CVֵ��
2.5��ͼ���ͼ������
2.5.1��ͼ���ͼ�����Ľ���
��ͼ����Ϊ��������ȷ�Ժ;��ܶȵ������£��ܹ����Ŀ�������˻�����DNA���Ũ�ȡ�
�ɲ����ݶ�Ũ�ȵ��˻�����DNA�������ж���ظ���⣬ȷ��95%�����ˮƽ�����˻�����DNA���Ũ�ȣ���Ϊ��ͼ���ޡ�ϵ��ϡ�Ͷ�Ӧ�ܹ����Ǵּ���������䣨0��100%�����ɸ��ݸ�Ũ���ݶȼ����ֱ���ж���Ҳ��ͨ���ʵ���ģ�ͣ���Probit�������������������������걨��Ʒ���趨�����µļ���ޣ�һ���ڸü��Ũ����Ӧ����95%�ļ���ʡ�
ͬʱ����������Ӧ���ۿ�ȷ������˻�����DNAŨ�����ޣ����ʵ������ˮƽ�µ��˻�����DNA���Ũ�ȡ�
Ӧ�����ٴ��������м������������о���
2.5.2��ͼ���ͼ��������֤
ѡȡ�뽨����ͬ���ٴ�����������֤���������ͼ���ͼ��������Ũ��ˮƽ��������������20�ε��ظ���⣬Ӧ����95%���ϼ����Ҫ��
2.6 ����������
2.6.1��������
��ͨ�����ٴ����������Ӹ������ʵķ�ʽ����������ǰ��������ʶл������Ӱ�����о�������Ӧ��������������������������������������������ϸ��Ũ��ˮƽ��������
����ע����������ÿ�ָ������ʵ�DZ�����Ũ�ȣ�������������������½������ۣ����и��ţ�Ӧȷ�����������ŵ����Ũ�ȡ�
Ӧ��Կ��ܵ���Դ����Դ�Ը�������и��������о�������ȫѪ��������Դ�Ը�������Ӧ����Ѫ�쵰�ס����������������ء����ס�����Դ�Ը�������Ӧ�������������Ҵ�����ȩ��Ԥ����Ⱥ��������ҩ���������ʹ�������ɡ��������������������� ���������� ˫�ȷ����ơ� ���������� ������ͪ�� ����ҡ� �������������ǰ��������
ͬʱ�����д����ԵĻ���������ͨ�����걨��Ʒ�벻�ܸø�����Ӱ��IJ���������������жԱȣ����и��������о���
2.6.2���淴Ӧ
Ӧ����л���������������ͬԴ�ԡ������淴Ӧ������HLA�����Ļ������н��н��淴Ӧ�о���
����ȫѪ���������ṩѪԴ�Ը�Ⱦ�����IJ�ԭ�壨�磺�ҸΡ����Ρ�HIV��÷�����Ľ��淴Ӧ�о���
2.7 ���������о�
2.7.1���걨��Ʒ�ļ��б�Ϊ������������Ҳ��ܷ��ͣ����������о���Ӧ���걨��Ʒ���Ƶ�ÿ�ֻ������ͽ����о�����֤��
2.7.2�������о�
���걨��Ʒ�ļ��б�ΪHLA-B27����Ӧ֤���걨�Լ����м����ͬHLA-B27���͵�������Ӧ���ٰ����й���Ⱥ��֪��������B2702��B2704��B2705��B2707�ȡ�
�������о�Ӧ�ֱ�������������ͽ��о��ܶȣ��ظ��ԡ��м侫�ܶȺ������Ծ��ܶȣ��о�����ͼ������֤��
2.8������ȡ/��������
�ڽ��к�����֮ǰ�������к�����ȡ/�������衣�����ʹ�õ����к�����ȡ�Լ�������ȡ���ᴿ�ȡ�Ũ�ȡ���ȡЧ�ʵ��о������������Ϻõĺ�����ȡ�Լ�����ƽ�бȶԡ�
����Ʒ�������ֻ����Ϻ�����ȡ�Լ�����ÿһ�ֺ�����ȡ�Լ�������ϼ���Լ����о��ܶȡ�����Ϳ����ŵ���֤��
2.9��Ӧ��ϵ�о�
2.9.1�����ɼ��ʹ���
�����������IJɼ���������ʽ����ȷ�����ı�����ʡ�
2.9.2��Ӧ��ϵ
Ӧ���ݲ�Ʒ���ԣ��ṩ��Ӧ��ϵ��ȷ���о����ϡ������������ں�����ȡ�õ����������ϴ�������PCR��Ӧ���������Լ��ļ������������øŨ�ȡ�����/̽��Ũ�ȡ�dNTPŨ�ȡ�������Ũ�ȼ���Ӧ�������磺�¶ȡ�ʱ�䡢ѭ�������ȡ�
���ж�����û��ͣ����ṩ��ͬ���û��ͻ��ߺ���ֵ��ȷ�����ϡ���ͬ���û��͵ķ�Ӧ��������в���Ӧ�ֱ����������ύ��֤���ϡ�
3.�ȶ����о�
�����˿ɸ���ʵ����Ҫѡ��������ȶ����о��������ȶ����о�����Ӧ�����о�������ȷ�����ݡ���������鷽�����������ݡ�����Լ����ۡ�
3.1ʵʱ�ȶ����о�
�ύ���������걨��Ʒ��ʵ�ʴ��������±�������Ʒ��Ч�ں���о����ϡ���ȷ����Ļ������������¶ȡ�ʪ�Ⱥ��գ�����Ч�ڡ�
3.2ʹ���ȶ���
�ύ�걨��Ʒʵ��ʹ���ڼ��ȶ��Ե��о����ϣ�Ӧ����������ɳɷֵĿ����ȶ��ԡ�����ʱ�ύ�����ȶ��ԡ������ȶ��Լ����ڴ����о����ϵȡ���ȷ��Ʒʹ�õ��¶ȡ�ʪ�������ȡ�
3.3�����ȶ���
�����ֽ��ͼʾ�ķ�ʽ��ϸ���������װ����ȷ��Ʒʵ������Ļ������������¶ȡ�ʪ�ȡ����պͻ�е�����ȣ�����¶����������������ύ�걨��Ʒ�����ض�����Ԥ�ڵ�������������о����ϡ�
4.�����ж�ֵ�о�
�����ж�ֵ��Ϊ�ܹ��������ļ��ȷ�Ե��ٽ�ֵ��Cut-off�����о�����ӦΪԤ��ʹ����Ⱥ����ʵ�����������й���Ⱥ����HLA-B27������������ԴӦ���е������������ԣ����Dz�ͬʱ�������/����״��
���������߹���������ROC�����ߵķ�ʽ�����о�����ɲ���������ѧ�����ķ������������ж�ֵ�о���
������Ӧ�������鷽�������������Լ����������Աȷ�������������������������ύ�����ж�ֵ�о�����������������Ϣ�б������ٰ������塢�Ա����䡢��Դ��Ψһ����Դ��š��ٴ������Ϣ�ȣ����������ȡ�
����Լ��ж����ڻ�����Ӧ����˵��������Χ��ȷ��������
�ṩ�ڱ�������Χ��ȷ���������о����ϡ�
5.��������
5.1��Ҫԭ�����о�����
�����Ʒ����Ҫԭ���ϰ������̽�롢ø��dNTP���������/����������ʿ�Ʒ���ο�Ʒ�ȡ�Ӧ�ṩ��Ҫԭ���ϵ�ѡ����Դ���Ʊ����̡��������Ʊ�������о����ϡ�����Ҫԭ����Ϊ��ҵ���ƣ�Ӧ�ṩ��ϸ���Ʊ����̼�֧�������ϣ�����Ҫԭ����Դ�����Ӧ�ṩ���ϰ�����ѡ���ԭ���ϵ����ݼ��Ա�ɸѡ�������ϡ��������ṩ�����������������鱨�棬�Լ���ԭ���ϵ�����������������ϡ�
5.1.1�����̽��
Ӧ�������̽������ԭ���ṩ����̽�����С��л������м����ߵĶ�Ӧ�����������������������̽���Թ�ɸѡ����Դ���λ��ļ�������Ⱥ������ԵȽ������ۣ�ѡ���������̽����ϣ����ύ��ϸ��ɸѡ�о����ݡ�ͬʱӦ������̽�뼰���������빫�����ݿ����ͬԴ�Է���������ͬԴ����Ӧ���������Ƿ���н��淴Ӧ��
������Ӧ���ѡ�������̽��ԭ���Ͻ����������ۣ�һ���������ۡ������������ȡ�Ũ�ȡ�̽��ӫ���ǻ��ŵļ��������ͷ��䲨�����Լ�����������ȣ����������۽��������������������
5.1.2������������գ�dNTP��
����dATP��dCTP��dGTP��dTTP��dUTP��Ӧ�ύ���䴿�ȡ�Ũ�ȡ������ȶ����Լ���������������ϣ���ȷ����������
5.1.3ø
�����漰����ø����DNA�ۺ�ø�������DNA�ǻ���ø�ȣ�Ӧ�ֱ��ø���ԡ����ȶ��ԡ�����������Ƚ������ۺ���֤����ȷ����������
DNA�ۺ�øӦ����DNA�ۺ�ø���ԣ���������ø���ԣ������ȶ��ԡ�UDG/UNGӦ����ˮ����������ռ��Ļ��ԣ���������ø����������ø���ԡ�
5.1.4�ʿ�Ʒ
ѡ���ⷶΧ��һ�ֻ���������Ʊ������ʿ�Ʒ��ͬʱ���ò�����������еĿհ��ʿ�Ʒ���ڽ�����Ⱦ���ʿء�
HLA-B������ڸ�����죬�����������������HLA-B27������Լ����������������ڱ���ա�
�ʿ���ϵӦ�ܹ��Լ��ȫ���̽�����Ч���������ƣ������Լ����������ܡ����ܵ�������Ӧ������������ƣ���������Ⱦ��������ɵļ����Ի�����Խ�����ʿ�Ʒ�ɲ����ٴ�����������ȡҺ��ϸ��ϵ�ȡ��հ��ʿ�ƷӦ�������������ƽ����ȡ��������Ӧ����ʿ�Ʒԭ����Դ��ѡ���Ʊ�����ֵ���̵��ṩ��ϸ���о����ϣ������ʿ�Ʒ�ļ����������ȷ�ķ�ΧҪ��
5.1.5 �ڱ�
�ڱ꣬�ֳ��ڶ��գ��ɶԹ������Ƶ��µ����Խ�������������ƣ�Ӧ��к���һͬ��ȡ������������������ڱ�����̽����ƺ���ط�Ӧ��ϵ��Ũ������ȷ��֤����Ҫ��֤�ڱ�ӫ��ͨ�������Ե�����������Ҫ�������Ͷл�������ɵ����ơ���ȷ�ڱ�ļ����Ctֵ��Χ�������ѧ�����ڱ꣬�Դ���������ȡ���������Լ��ķ�Ӧ��ϵ���м�ء�
5.1.6������ȡ/�����Լ������У�
Ӧ�ṩ�Լ�����Ҫ��ɡ�ԭ�����ܼ���ص�ɸѡ����֤���ϡ�
5.1.7��ҵ�ο�Ʒ
�����Ʒ����ҵ�ο�Ʒһ��������Բο�Ʒ�����Բο�Ʒ������ο�Ʒ�;��ܶȲο�Ʒ����ҵ�ο�ƷӦΪ��ʵ��������Ӧ���ݲ�Ʒ�ص��������֤��ʵ����Ҫ�������á�
Ӧ�ύ��ҵ�ο�Ʒ��ԭ����Դ��ѡ���Ʊ���Ũ�ȼ���������ȷ�Ϸ������Լ�������о����ϡ���ҵ�ο�Ʒ�����ý������£�
���Բο�ƷӦ�����ɼ���ĸ�HLA-B27��������Ӧ���ò�ͬŨ��ˮƽ��
���Բο�ƷӦ���Ǽ�������Ե����ۣ�����ͬԴ���н��淴Ӧ����������������HLA-B27���������ȡ�
����ο�Ʒ�����������Բο�Ʒ��ͬ��Ũ��ˮƽӦ������ͼ����Ũ�Ȼ��Ը�����ͼ����Ũ�ȡ�
���ܶȲο�Ʒ���ٰ���HLA-B2704��HLA-B2705��ÿ�����������ߡ�������Ũ��ˮƽ������һ��Ũ��ӦΪ���������Ũ�ȡ�
5.2�������������
�����Ʒ���������о�������Ҫ��������Һ���ƣ�����̽��Ũ�ȡ�øŨ�ȡ�dNTPŨ�ȡ�����Һ����Ũ�ȵȣ�����װ�Ͷ��ɡ�ӫ���ǵȹ��չ��̵�������ȷ�����ݡ�
��������Ӧ�Թؼ�����������Ч���ƣ��ɽ�Ϲ�������ͼ�����������չ��̣���ȷ�ؼ������ʿز��裬����ϸ˵���ò�����ʿط������ʿر������ύ��Ҫ�������յ�ȷ�����Ż��о����ϡ�
���ģ��ٴ���������
�ٴ�����Ŀ�չ���������ƶ��Լ������д�Ⱦ�Ӧ������ط��漰����������Լ��ٴ����鼼��ָ��ԭ������ҩƷ�ල������ͨ��2021���72�ţ���Ҫ������ط��桢�ļ��и��£��ٴ�����Ӧ���ϸ��º��Ҫ����
1.�ٴ��������
Ӧѡ������3�ң���3�ң��ѱ������ٴ����������������ط����Ҫ��չ�ٴ����顣������Ӧ���ݲ�Ʒ�ص㼰Ԥ����;���ۺϲ�ͬ�������ֺ����в�ѧ����������ѡ���ٴ�����������ٴ��������Ӧ���з�������ѧ�����������ƣ�ʵ�������ԱӦ���㹻��ʱ����Ϥ���ϵͳ�ĸ����ڣ���Ϥ�ٴ����鷽����
2.�ٴ����鷽��
���걨��Ʒ����ͬ���Ʒ���У�����������ѡ�����������е�ͬ���Ʒ��Ϊ�Ա��Լ�/�����������걨��Ʒ��֮���жԱ������о��������걨��Ʒ���ٴ����ܡ��Ա��Լ���ѡ��Ӧ��Ԥ����;������Ҫ������ܵȷ��棬ȷ�������걨��Ʒ���нϺõĿɱ��ԡ���Ա��Լ����ɷ��ͣ���Ӧѡ���ٴ��ο��������磺һ�������ٴ������е������������з��ͣ�ȷ���걨��Ʒ�����ƵĻ������;�����֤��
3.������ѡ�����������
3.1������ѡ��
������ӦΪǿֱ�Լ��������ƻ��ߣ�Ӧ�����������֢״�������ȣ��磺��������ʹ�����ܹؽ�����ֺ�ȣ��ٴ����黹Ӧ���벿����Ҫ������ϵ��������ߣ��磺���ʪ�ؽ���ϵͳ�Ժ���Ǵ��Ļ��ߵ�֢״������Ⱥ��
3.2 ��������
���õ���������һ��Ϊ����ȫѪ���ٴ�����Ӧ�����ٴ�ԭʼ��������Ӧֱ�Ӳ�����ȡ�Ļ�����DNA�������顣�ٴ������IJɼ����������������ȡ��Ӧͬʱ�����걨��Ʒ˵�����Լ��Ա��Լ�˵���飨�����ã������Ҫ��
4.�ٴ�����������
�ٴ�����������Ӧ����ͳ��ѧҪ�ɲ����ʵ���ͳ��ѧ�������й��㣬ͬʱӦ���㷨�������������Ҫ��
������õ���Ŀ��ֵ���������������㡣���Է����ʺ����Է����ʵ��ٴ��ɽ��ܱ���P0�������������95%��������ָ��PT�ӽ�100%ʱ�������������㷽�����ܲ����ã�Ӧ����ѡ��������˵ķ������������������ͳ��ѧ�������羫ȷ���ʷ��ȡ�
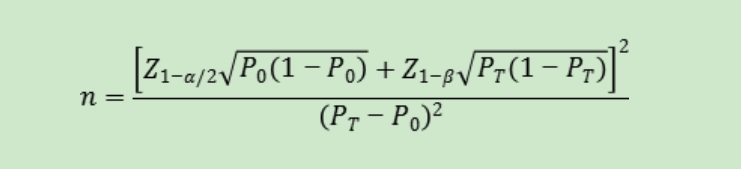
������ʽ�У�nΪ��������Z1-��/2��Z1-��Ϊ������ˮƽ�Ͱ��նȵı���̬�ֲ��ķ���λ��P0Ϊ����ָ����ٴ��ɽ��ܱ���PTΪ�걨��Ʒ����ָ��Ԥ��ֵ��
�ٴ���������������ȷ��ʱӦ�������������������������������Ļ����ϣ�ͬʱ������������������������������Լ�������Ҫ����ĸ������������淴Ӧ����������ʵ���������������.
���걨��Ʒ�ļ��б�Ϊ������������Ҳ��ܷ��ͣ��ٴ�����Ӧ���걨��Ʒ���Ƶ�ÿ�ֻ������ͽ�����֤��ÿ�ֻ������;�Ӧ��һ��������������
���걨��Ʒ�ļ��б�ΪHLA-B27�����ٴ���������Ӧ�Գ����������ͽ�����֤������Ӧ��֤HLA-2704��HLA-2705��HLA-2702��HLA-2707��,ÿ�ֻ������;�Ӧ����һ��������������
5.ͳ��ѧ����
Ӧ��������Ⱥ�����˿�ѧ�������������䡢�Ա���ٴ���ϱ�����Ϣ�ȡ�Ӧ�ܽ���������������������2��2���ֱ��ܽ������Լ��Ķ��Լ���������ֱ�������������ʡ����Է����ʺ��ܷ����ʼ���95%�������䡣���ڿɷ��͵��Լ���Ӧ��2��2���ֱ��ܽ������Լ���ͬ�������͵����Լ���������ֱ������ͬ�������͵����������ܷ����ʼ���95%�������䡣
�������ռ������У����������Լ��������һ�µ�������Ӧ���ú����������и��ˣ����Բ���ԭ����з����������踴�ˣ�Ӧ˵�����ɡ�
6.����ѧҪ��
�ٴ����������Ϻն��������Ե�����ѧ���о���Ӧ�����ٴ������������Ļ�ú��������������ߵķ��գ���������ίԱ����飬���������ίԱ���ͬ�⡣ע���걨ʱӦ�ύ����ίԱ�����������
7.�ٴ����鷽��
���ٴ���������ķ�������Ӧ����һ�£��ұ�֤�������ٴ������������ѭԤ���ķ�������������Ķ��������������Ӧ���ٴ����������ʵ�����ڲ��ɱ�ʵ���ҵļ�����Ա������ɣ��걨��λ�ļ�����Ա�����б�Ҫ�ļ���ָ���⣬�����������ʵ����̡�
���鷽��Ӧȷ���ϸ����ѡ/�ų������κ�����ѡ���������ų����ٴ����鶼Ӧ��¼�ڰ�����ȷ˵��ԭ��������������̺ͽ���ж�ʱӦ����ä���Ա�֤�������Ŀ��ԡ����ٴ��������ѡ�õĶԱ��Լ�/����Ӧ����һ�£��Ա���к�����ͳ��ѧ������
8.��������
�ٴ����鿪ʼǰ����������ٴ������Ԥ���飬����Ϥ������������鷽���IJ������������������ܵȣ�����ȿ�������������������̶�Ӧ������Ч�����������£�����ȱ�֤�������ݵ�ȷ�Լ����ܶȡ�
9.�ٴ����鱨��д
�ٴ����鱨��Ӧ�ö������������Ƽ������ؼ�����������������IJ�����Ӧ�ö������ٴ�����ʵʩ���̡�������������۵Ƚ���������������������Ӧ������Ҫ�Ļ������ݺ�ͳ�Ʒ������������ó��ٴ�������ۡ��ٴ����鱨���д�ο�����������Լ��ٴ����鼼��ָ��ԭ�����Ҫ��
���壩��Ʒ˵����
��Ʒ˵�����ʽӦ���ϡ���������Լ�˵�����дָ��ԭ��Ҫ��Ʒ˵����ļ�������Ӧ��ע���걨�����е�����о��������һ�£���ijЩ���������Բο����ף�Ӧ�Թ淶��ʽ�Դ����ݽ��б�ע��������ע�����������Ϣ��
�������ݽ���HLA-B27�������Լ�˵������ص����ݽ�����ϸ˵����˵������������Ӧ���ݡ���������Լ�˵�����дָ��ԭ����Ҫ����б�д��
1.��Ԥ����;�� Ӧ���ٰ������¼��������ݣ�
1.1����Ʒ�������ⶨ�Լ��������������Ѫ�����е�HLA-B27����
1.2���ܱ����������ٴ�����֢�Ĺ�ϵ����ȷHLA-B27���й���Ⱥ�еķֲ�Ƶ�ʣ����ٴ�����֢��Ⱥ�е�����Ƶ����
1.3��ȷ����Ʒ����������ٴ��ο����ٴ�ҽ��Ӧ��ϻ��߲��顢��Ч������ʵ���Ҽ��ָ��ȶԱ���Ʒ�ļ���������ۺ��жϡ�
2.������ԭ����
2.1�Ա����л�����HLA-B�����е�����λ�ã�Ƭ�δ�С����������������ϸ���������Լ�������̽�롢������Խ�����ж���ʽ�Ƚ�����ϸ�Ľ��ܣ���ȷ�ڱ��������Ƽ������á��Բ�ͬ������Ӧ����ϡ��ʿ�Ʒ���ü�ӫ���źż��ԭ���Ƚ��н��ܡ�
2.2�練Ӧ��ϵ����������صķ���Ⱦ��֣���UNGø����ҲӦ�������û������м�Ҫ���ܡ�
3.����Ҫ��ɳɷ֡�
��ȷ�Լ����и���ֵ����ơ�������װ����
��ȷ������е����̽�롢ø��dNTP������Һ���ơ����������ơ�
��ȷ��Ҫ��δ�ṩ�IJ��ϣ����������ȡ�Լ��ȵIJ�Ʒ���ơ�ע���ˣ����ż�ע��֤�š�
��ȷ��ͬ�����Լ����и�����Ƿ���Ի�����
4.�����鷽����
��ϸ˵����������ĸ������裬������
4.1������������Ҫʱ��Ӧ��ȷʵ���ҷ��������黷�����¶ȡ�ʪ�ȺͿյ�����������Ƶ�ע�����
4.2�Լ����Ʒ�����ע�����
4.3����������ȡ���������������輰ע���������ȫѪ����������ϴ������ȣ��Ժ�����ȡ�������ڽ��к������������ƣ���ȷ��ȡ�����Ũ�ȴ��ȵ�����Ҫ��
4.4������Ӧǰ�������������˳��ȡ�
4.5 PCR���ε��¶ȡ�ʱ�����á�ѭ�������û���Ӧ���Զ������������ע�����
4.6�������ã����������̽���ӫ���ر���������ӫ��ͨ��ѡ��ȡ�
5.�������ж�ֵ��
��ȷ�л�����ڱ����������ж�ֵ����Ҫ˵����������֤�����ж�ֵ�Ļ�����Ϣ����������������Ⱥ���������塢�Ա����䡢����/����״̬�ȣ��Ͳ��õ�ͳ��ѧ������
6.���������Ľ��͡�
���б���ʽ�������п��ܳ��ֵĽ������Ӧ�Ľ��͡�
7.�����鷽���ľ����ԡ�
7.1�����ɼ���HLA-B27���ͣ���ȷ��������δ������֤��
7.2��ȷ���ܳ��ֵĽ��淴Ӧ�������ɼ����ͬԴ���С�����HLA����ȡ�
7.3�йؼ����Խ���Ŀ����Է�����
7.3.1�������������ɼ������ͼ������������Ƚ�����п��ܵ��¼����Խ����
7.3.2δ����֤���������Ż�PCR�������ӵȿ��ܻᵼ�¼����Խ�������У���
7.4 ��ȷ�Ƿ������HLA-B27�Ӻϻϡ�
7.5��ȷ������Ⱥ��Ҳ�ɱ���HLA-B27���ٴ�ʹ����Ӧ��ϼ����������֢״/��������ʷ��ʵ���Ҽ�鼰���Ʒ�Ӧ������ۺϿ��ǡ�
8.����Ʒ����ָ�
8.1���Ϲ��ұ�Ʒ�������
8.2ȷ���о������ͽ����
8.3���ܶ��о����ܽᣬ����ʵ�鷽����������������Ũ��ˮƽ�������ܶȿ������أ�ʱ�䡢�ص����Ա�ȣ���
8.4��ͼ���ޡ���ȷ�ɼ��DNAŨ��ˮƽ��Χ��
8.5�����о��������ȷ����Դ����Դ�Ը�����Լ����������Ӱ������Ũ�ȡ�
8.6���淴Ӧ�о��������ȷ�����о���ͬԴ����/HLA�����Ƿ���ڽ��淴Ӧ��
8.7 �������о��ܽᡣ��ȷ�ɼ�������͡�
8.8�ٴ������ܽᡣ
9.��ע�����Ӧ���ٰ����������ݣ�
9.1��ò�Ʒ������Դ����Դ�����ʣ�Ӧ��������DZ�ڸ�Ⱦ�Եľ��档
9.2�ٴ�ʵ����Ӧ�ϸ��ա�ҽ�ƻ����ٴ���������ʵ���ҹ����취�����йط�������ѧʵ���ҡ��ٴ���������ʵ���ҵĹ����淶ִ�С�
����������������ϵ�ļ�
������Ӧ��������ע��ʱ�ύ���Ʒ���ơ������йص�����������ϵ������ϡ�������Ʒ���������̣��ṩ������������ͼ����ȷ�걨��Ʒ��Ӧ�����ԭ�����̣�������Ҫ���Ƶ�����Ŀ����Ҫԭ���ϡ��ɹ�������Դ���������Ʒ�����
�����ã�Ӧ���ṩ��˲��Ʒ�������ͨ���˲��Ʒ�������������������յȷ���ĶԱ�˵����