

| ��ָ��ԭ��ּ��ָ��ע�������˶Ը��廯��ҩ�������Լ��ٴ��������ϵ�����д��ͬʱҲΪ�������������ṩ�ο�����ָ��ԭ���ǶԸ��廯��ҩ�������Լ��ٴ����۵�һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ���� |
��ָ��ԭ��ּ��ָ��ע�������˶����廯��ҩ��������Լ��ٴ���������������д��ͬʱҲΪ�������������ṩ�ο���
��ָ��ԭ���Ƕ����廯��ҩ��������Լ��ٴ�������һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����ã��������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ����
��ָ��ԭ���ǹ�ע�������˺ͼ��������Ա��ָ�����ļ����������������������漰������������Ϊ����ǿ��ִ�У�Ӧ����ѭ��ط����ǰ����ʹ�ñ�ָ��ԭ��������ܹ�������ط���Ҫ�������������Ҳ���Բ��ã�����Ҫ�ṩ��ϸ���о����Ϻ���֤���ϡ�
��ָ��ԭ���������з���ͱ���ϵ�Լ���ǰ��֪ˮƽ���ƶ��ģ����ŷ���ͱ��IJ������ƣ��Լ���ѧ�����IJ��Ϸ�չ���������Ҳ����ʱ���е�����
һ�� ���÷�Χ
��������ҩ�������ѧ�õ���Ѹ�ͷ�չ��Խ��Խ���ҩ������������������ӿ�֣��Ƚϵ��͵ı�־���磺ҩ���лø����ҩ�����ðе�Ļ�����졣
ҩ���лø��ҩ�����ðе�Ļ�������ͨ��Ӱ��ҩ�������Ũ�ȺͰ���֯��ҩ��������ԣ�����ҩ�ﷴӦ����ҩ����Ч��ҩƷ������Ӧ���ĸ�����졣ҩ������������־��ļ�⣬�����廯��ҩ�����⣬���ٴ�ʵʩ���廯ҩ�����Ƶ�ǰ�ᡣ
���廯��ҩ������Ŀ����Ҫ�����������棺
1. ���ݸ����Ŵ���ϢԤ��ҩ����Ч���Ӷ�������ҩ�����������ҩ���࣬�Ը���ҩ����Ч��
2. ���ݸ����Ŵ���ϢԤ��ҩ�ﲻ����Ӧ���Ӷ�������ҩ�����������ҩ���࣬�Ա�������ҩƷ������Ӧ��
��ָ��ԭ���������廯��ҩ�������Լ���ָҩ���лø��ҩ�����ðе����������Լ������������û����ⷽ��������������л������ض����죬�Ӷ�ָ���ض�ҩ��ʹ�õļ���Լ���
��ָ��ԭ�������ڰ�������Լ���
��ָ��ԭ�������ڽ�����������Լ���Ʒע������ͱ��ע������IJ�Ʒ���ص�������廯��ҩ��������Լ��ٴ���������������д��ȷҪ��
����ע�����Ҫ��
��һ�� ���廯��ҩ�������Լ�������켰���ҩ���ѡ��
���廯��ҩ��������Լ��ڲ�Ʒ�������ؼ������������ض�������켰��ָ�����ض�ҩ����ѡ��
�������Լ���������������У�������Ӧ��ֿ��DZ�ѡ��������ҩ����ٴ�������ٴ��Ͽɶȡ��Լ���ѡ�����������������й���Ⱥ�ķ���Ƶ�ʵȣ���ֵ��й�����ҩƷ˵���顢Ȩ�����ݿ⣨�磺PharmGKB���ݿ⡢CPIC���ݿ��DPWG���ݿ⣩�����ָ�ϣ�ѡ���ʵ�����������ҩ����ϡ�
Ӧ�ر����ѵ��ǣ�
1. ������Ӧ�ر��ע����й���Ⱥ���о����ݣ���������ҩ�����Ӧ�ʺ��й���Ⱥ���ص㣬�����й���Ⱥ���ٴ�ʹ����Ҫ��
2. �����ҩƷ������Ӧ��صĸ��廯�������Լ������ٴ������ȷ�ϣ���Ҫ��������ָ�ϡ�Ȩ�����ݿ⡢ҩƷ˵������ٴ��о�֤�ݵȡ��ڿ��������Լ�ʱ�����������˽��г�ֵ��к�ѡ����������ҩƷ������Ӧ��ϵ��ȷ�Ļ����ҩ����Ͻ��в�Ʒ�з���
3. ��Ը��廯��������ϼ���Լ����ڲ�Ʒ��ƿ���ʱ��Ӧ��ֿ��Ƕ��������ʱ����ָ���ĵ����������ҩ���ھ���������Ⱥʱ�����ɿ���������ϡ�
���������廯��ҩ�������Լ��ٴ�����Ҫ��
1. �ٴ���������
�����Լ����ٴ����۰��������������ݣ�
1.1��Ʒ�ٴ������ȷ�ϣ������������ҩ�ﷴӦ��ȷ�ϣ���ͨ������ָ�ϡ�Ȩ�����ݿ⡢ҩƷ˵������ˮƽ�ٴ��о�֤�ݵ��ṩ�ٴ�֤�ݡ���Ʒ�ٴ�����ȷ�ϵ�֤�ݣ���Ϊ����֤�����͵���ϡ�
1.2��Ʒ�ٴ�������ܵ�ȷ�ϣ�Ӧͨ���ٴ������ṩ�ٴ�֤�ݡ�
1.3 ��ͨ���ٴ������ṩ�ٴ�֤�ݣ�Ӧע�⣺
1.3.1�����ٴ�����Ӧ���ϡ���������Լ�ע���뱸�������취������ҽ����е�ٴ��������������淶��������������Լ��ٴ����鼼��ָ��ԭ�������з�����Ҫ��
1.3.2��Ϊ�����ٴ��������ݣ���Ӧ�������������ֲ�����ٴ���������Ӱ�죬���б�Ҫ��Ӧ���ݡ�ʹ����������Լ������ٴ��������ݵ�ע�����ָ��ԭ��������ٴ�֤�ݡ�
2. ��Բ�Ʒ�ٴ�������ܽ��е��ٴ�����
�����˵�������Լ��ٴ�������Ӧ��ע���ص����⡣
2.1 �ٴ��������
�ٴ�����Ӧ�ڲ�����3�ң���3�ң����߱���Ӧ�����Ұ��涨������ҽ����е�ٴ����������չ��
2.2 �Աȷ���
�����й���������ͬ���Ʒ���е��Լ�����ѡ���й����������еġ����пɱ��Ե�ͬ���Ʒ��Ϊ�Է���������þ��пɱ��Ե��ٴ��ο��������磺һ������ȣ���Ϊ�Աȷ������бȶ��о����������й���ͬ���Ʒ���е��Լ���������ѡѡ��ͬ���Ʒ��Ϊ�Աȷ�����
�����й���������ͬ���Ʒ���е��Լ��������걨��Ʒ����пɱ��Ե��ٴ��ο��������磺һ������ȣ���Ϊ�Աȷ�����
�Աȷ����Ŀɱ���Ӧ��ע�������������͵Ŀ����ԡ�Ԥ����;��������Ⱥ�����������켰ָ��ҩ��Ŀɱ����Լ���Ʒ����ָ��Ŀɱ��Եȡ�
2.3 ������ѡ��
�ٴ������������ȺӦ�����걨��Ʒ��Ԥ��������Ⱥ��ӦΪ��ָ��ҩ���Ԥ��ʹ����Ⱥ���������걨��Ʒ��Ԥ����;�����������Ⱥ�����ٴ��������ٴ����鷽��Ӧ��ȷ����������������˵�����ݡ�����������Ӧ�ܹ�������Ʒ������Ⱥ�ĸ������Σ����磺Ӧ������ͬ���䡢�Ա������ߣ����漰�����Ӧ֤��ÿ����Ӧ֤���߾�Ӧ����һ��������
2.4������
�ٴ�����������Ӧ�����ʵ���ͳ��ѧ�������й��㣬����ϸ������ʹ��ͳ�Ʒ�������������ȷ�����ݡ��˲����ٴ�����Ŀ��Ϊ�������ּ�ⷽ��֮���һ���ԣ�������õ���Ŀ��ֵ����������������Ĺ��㡣ͨ�����Է����ʺ����Է��������ֱ����������������������������������Ӧ���������ÿ������λ���ֱ�����������Ĺ��㡣
���ÿ������λ�㣬�������Է����ʵ��ٴ��ɽ��ܱ���P0��һ�㲻����95%��������ָ��PT�ӽ�100%ʱ�������������㷽�����ܲ����ã�Ӧ����ѡ��������˵ķ������������������ͳ��ѧ�������羫ȷ���ʷ��ȡ�
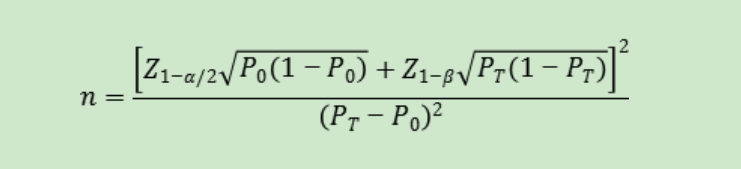
��ʽ�У�nΪ��������Z1-��/2��Z1-��Ϊ������ˮƽ�Ͱ��նȵı���̬�ֲ��ķ���λ��P0Ϊ����ָ����ٴ��ɽ��ܱ���PTΪ�걨��Ʒ����ָ��Ԥ��ֵ��
���ÿ������λ�㣬�����������������ӦΪ����ͻ���ͺ��Ӻ��͵��ܺͣ�Ҫ��ͻ���ͺ��Ӻ��;�Ӧ����һ�����������紿��ͻ���ͻ��Ӻ������������ٴ�����Ƶ�ʵͽ��ѻ�ã��ɻع����ٴ�ԭʼ�������и��������ھ�����ȷ�ٴ�����ı���λ�㣬��ȷ������ͻ���ͣ�Ӧ�ṩ֤�ݣ����ڸñ���λ��������������������Ҫ���ǰ���£���������ٴ���ͻ���ͻ��Ӻ��͵������������ٴ�������δ��ȷ���µı���λ�㣬�ɲ��ûع����ٴ�ԭʼ���������ٴ��о�����֤����ͻ���ͻ��Ӻ�ͻ�������۵ij���ԡ�
�ٴ�������������ȷ��ʱӦ�������������������������������Ļ����ϣ�ͬʱ�����������������������������������ʵ������ʵ�����������������
2.5 ͳ�Ʒ���
Ӧ����������Ⱥ�����˿�ѧ�����������Ա����䡢�������ͺͻ����������ܽ����
���ÿ������λ�㣬һ����3x3�����ֱ��ܽ��걨��Ʒ��Աȷ����Ķ��Լ�������������ַ��������Է����ʣ���������ͻ���ͺ��Ӻ������壩�����Է����ʣ�Ұ���ͣ�������ͻ���͵������ʡ��Ӻ��ͷ����ʡ�Ұ���ͷ����ʡ��ܺͷ���������Ӧ��95%�������䡣
��Բ�һ��������Ӧ���ú����ķ�������ȷ�ϣ��Բ�һ�µ�ԭ���ۺϽ��з�����
3. ��Բ�Ʒ�ٴ����壨�ٴ���Ч�ԣ�ȷ�ϵ��ٴ�֤��
3.1 ����ͬ���Ʒ���е��걨��Ʒ
�������й�����ͬ���Ʒ���е��걨��Ʒ������Ϊ���ٴ������ѵõ��Ͽɣ������ٽ����ٴ���Ч�Ե�ȷ����
3.2��ͬ���Ʒ���е��걨��Ʒ
3.2.1 ��������λ����ҩ�ﷴӦ�Ĺ�ϵ���ڹ����������ָ���������ҩƷ˵�����б���ȷ˵�������Ҿ��нϸߵ�֤�ݼ������漰֤�ݼ��𣩣�����Ϊ���ٴ������ѵõ��Ͽɣ������ٽ����ٴ���Ч�Ե�ȷ����
3.2.2 ��������λ����ҩ�ﷴӦ�Ĺ�ϵ���ڹ������ָ����Ȩ�����ݿ�����ҩ��˵�����б���ȷ˵�������нϸߵ�֤�ݼ������漰֤�ݼ������Ұ�����ֵ��й���Ⱥ���ٴ�����������Ϊ���ٴ������ѵõ��Ͽɣ������ٽ����ٴ���Ч�Ե�ȷ����
3.2.3 ��������λ����ҩ�ﷴӦ�Ĺ�ϵ���ڹ������ָ�ϡ�Ȩ�����ݿ�����ҩ��˵���鱻��ȷ�����Ҿ��нϸߵ�֤�ݼ������漰֤�ݼ��𣩣��������Ͽɲ�δ�����й���Ⱥ���ٴ����ݣ�������Ӧ������۹��������ֵIJ���Ա���λ����ҩ�ﷴӦ��ϵ��Ӱ�죬���б�Ҫ��Ӧ�ύ�����й���Ⱥ���ٴ�֤�ݡ�
3.2.4��������λ����ҩ�ﷴӦ�Ĺ�ϵ��δ�ھ���������ָ�ϡ�ҩƷ˵�����Ȩ�����ݿ�ȷ������Ͽ��������˿�ͨ�����·�ʽ֤������λ����ҩ��Ĺ�ϵ��
3.2.4.1 ͨ������ѭ֤ҽѧ֤�ݡ�������Ӧ����ϵͳ���۵ķ�����ȷ���������ٴ����⣬���ƶ���ѧ�ļ���·���ؼ��ʡ�������Ч������ɸѡ�����ݴ����ȣ��Ը�ˮƽ�ٴ��о����ݽ���������ϵͳ���ۣ����ṩϵͳ���۱��档Ӧ����ע����ǣ��ò��ֽ��۵Ļ��Ӧ�ǻ��ڸ�ˮƽ�о����ݻ�õá�
3.2.4.2 ͨ�������ٴ�����
3.2.4.2.1 �ٴ��������
�ٴ�����Ӧ�ڲ�����2�ҡ��߱���Ӧ�����Ұ��չ涨������ҽ����е�ٴ����������չ
3.2.4.2.2 �ٴ��������
Ӧ�����ٴ�ԭʼ���������걨��Ʒ�ļ�⣬��ǰհ�Ի�ع����ռ��ٴ�����ʹ�����ҩ����ٴ���֣��磺��Ч������Ӧ�ȣ��������ض�����λ�㲻ͬ���������ٴ���ֵĹ�ϵ�����ո����ض�����λ�㲻ͬ�����Ϳ�ָ��ҩ��ʹ�õĽ��ۡ�����ûع��Է�ʽ��Ӧ��������һ��ʱ���ڵ���ػ��ߣ����ý��в�����ѡ��
�ٴ���ֱ���ָ����趨Ӧ�г�ֵ����ݣ�Ӧ���ٴ�������������ȷ�趨�����ݣ����ṩ��ֵ�֧�����ϡ���ҩƷ������Ӧ��صIJ�Ʒ���ٴ���ֱ���ָ�����Ϊ�Ƿ���ij�ֲ�����Ӧ�����ܹ����������Ƿ���������Ӧ�ļ��ָ�ꡣ��ҩ����Ч��صIJ�Ʒ���ٴ���ֱ���ָ�����Ϊ�Ƿ���Ч�����ܹ����������Ƿ���Ч�ļ��ָ�ꡣ
�ò����ٴ������걨��Ʒ�ļ�������ɲ����걨��Ʒ�ٴ��������ȷ��ʱ�ļ������
3.2.4.2.3 ������ѡ��
�ٴ������������ȺӦ�����걨��Ʒ��Ԥ��������Ⱥ��ӦΪ��ָ��ҩ���Ԥ��ʹ����Ⱥ��ӦΪҩ��˵��������Ӧ֤�����������������ٴ����鷽��Ӧ��ȷ���������������˵�����ݡ�����������Ӧ�ܹ�������Ʒ������Ⱥ�ĸ������Σ����磺Ӧ������ͬ���䡢�Ա������ߣ����漰�����Ӧ֤��ÿ����Ӧ֤�Ļ��߾�Ӧ����һ��������
3.2.4.2.4������
�ٴ�����������Ӧ�����ʵ���ͳ��ѧ�������й��㣬����ϸ������ʹ��ͳ�Ʒ�������������ȷ�����ݡ��˲����ٴ�����Ŀ��Ϊ��������λ�������ҩ�������������ˣ����ÿһ����λ�㣬��ͬ�����͵IJ���������ͻ���͡��Ӻ��ͺ�Ұ���ͣ�Ӧ����һ�����������Ա������Ч��ͳ��ѧ������
3.2.4.2.5 ͳ�Ʒ���
Ӧ����������Ⱥ�����˿�ѧ�����������Ա����䡢�������ͺͻ����������ܽ�ȣ�ͬһ����λ�㲻ͬ�����͵���ȺӦ������ͬ���˿ڻ��ߡ�
���ÿ������λ�㣬Ӧ�ֱ������ͬ�����ͣ�����ͻ���͡��Ӻ��ͺ�Ұ���ͣ������ҩ�������ԣ�������ͳ��ѧ���������ս���Ӧ��֧�ֱ���λ�����Чָ���ض�ҩ���ʹ�á�
3.2.5 �����±���λ���ٴ������ȷ�ϣ���Ϊ��������֤�ݵ���ϡ�
4. �漰�ٴ�����ʱ���ٴ����鷽����С��ͱ���
���ٴ��������Ӧ����ͬһ�ٴ��������������������ٴ�����������ϸ�ִ�У���������Ķ�������Ӧ������������͡��Աȷ���ѡ��������ѡ������ָ�ꡢͳ�Ʒ����������������������������Ҫ���������ȷ�Ĺ涨�������ݸ��ٴ���������������ȷ������������ƻ���
���ٴ��������Ӧ���ٴ��������ݺ�ʵʩ��������ܽᣬ�����ٴ�����С�ᡣ
�ٴ����鱨��Ӧ�������ٴ�����ʵʩ���̡����������������۵Ƚ���������������������Ӧ������Ҫ�����ݺ�ͳ�Ʒ������������ó��ٴ�������ۡ��ٴ����鱨���д�ο�����������Լ��ٴ����鼼��ָ��ԭ�����Ҫ��
���ݻ��ܱ���Ӧ�ṩΨһ����Դ�������߱�š�������š������˿�ѧ��Ϣ���Ա�����ȣ����ٴ���ϱ�����Ϣ���������͡��걨��Ʒ���Աȷ����ļ��������Ϣ��
���漰ί�е������������вο��������ģ�Ӧ�ṩ�ٴ�������������������ǩ����ί��Э�顣ͬʱӦ�ṩ�ο���������ϸ���ϣ��磺����ԭ���������Լ����������ο�������������֤���ο������ʿء����͵�����ͼ�����ݵȡ���������Ӧ���ٴ��������ǩ��ȷ�ϡ�