

| ��ָ��ԭ��ּ��Ϊҽ����еע�������˽���Ӧ�ý�������߲�������ҩ�����ö��Լ���Լ���ע���걨�ṩ����ָ����ͬʱҲΪҽ����е�ල�������Ŷ�ע���걨���ϵ������ṩ�����ο�����ָ��ԭ���ǶԸ����Լ�ע���걨 |
ҩ�����ü���Լ�ע�����ָ��ԭ��
��2023�����棩
��ָ��ԭ��ּ��Ϊҽ����еע�������˽���Ӧ�ý�������߲�������ҩ�����ö��Լ���Լ���ע���걨�ṩ����ָ����ͬʱҲΪҽ����е�ල�������Ŷ�ע���걨���ϵ������ṩ�����ο���
��ָ��ԭ���ǶԸ����Լ�ע���걨���ϵ�һ��Ҫ��������Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����ã��������ã������������ɼ���Ӧ�Ŀ�ѧ���ݣ������ݲ�Ʒ�ľ������Զ�ע���걨���ϵ����ݽ��г�ʵ��ϸ������ע����������Ϊ�б�Ҫ���ӱ�ָ��ԭ�������о����ݣ������в��䡣
��ָ��ԭ���ǹ�ע�������˺ͼ���������Աʹ�õ�ָ�����ļ�����������ע���������漰������������Ϊ����ǿ��ִ�У�������ܹ�������ط���Ҫ�������������Ҳ���Բ��ã�����Ҫ�ṩ��ϸ���о����Ϻ���֤���ϡ�������ѭ��ط����ǿ���Ա���ǰ����ʹ�ñ�ָ��ԭ��
��ָ��ԭ���������з��桢����ϵ�Լ���ǰ��֪ˮƽ���ƶ��ģ����ŷ��桢���IJ������ƺͿ�ѧ�����IJ��Ϸ�չ����ָ��ԭ���������Ҳ����ʱ���е�����
һ�����÷�Χ
��ָ��ԭ������������������߲�������ҩ�����ý��ж��Լ���ҽ����;���Լ�������������⼼����������������İ붨��ҩ�����ü���Լ��ɲ��ձ�ָ��ԭ��Ӧ���ݲ�Ʒ�ľ�������ȷ�����������Ƿ����ã��粻���ã�Ӧ����ѡ��������������ѧ���Ե��о����輰��������ָ��ԭ�������ڽ����״�ע���걨��ע�����걨�IJ�Ʒ��
����ע�����Ҫ��
��һ�������Ϣ
1.��Ʒ���Ƽ��������
��Ʒ����Ӧ���ϡ���������Լ�ע���뱸�������취������ط����Ҫ���������Һ����Լ��У���������߲������������ݡ���������Լ�������Ŀ¼����������ҩƷ������ҩƷ��ҽ���ö���ҩƷ�����ص��Լ��������Ϊ�����࣬�������Ϊ6840��
2.������Ϣ
��������Ʒ�б��������ļ����걨ǰ���ܻ�������ϵ�����ͨ��¼�Լ��������������ļ���
��������������
����������Ҫ������������Ʒ������Ԥ����;���걨��Ʒ������ʷ��������˵�������ݡ����У���Ʒ������Ӧ�������ԭ������Ʒ��Ҫ�о�������ܽ�����ۡ���ͬ���/��ǰ����Ʒ�ıȽϵȡ���ͬ���/��ǰ����Ʒ�ıȽϣ�Ӧ���شӷ���ѧ������ԭ������Ʒ��Ҫ���ܵȷ�����ϸ˵���걨��Ʒ��Ŀǰ�г����ѻ���ͬ���Ʒ֮�����Ҫ����
��������Ӧ���ϡ���������Լ�ע���뱸�������취���͡����ڹ�����������Լ�ע���걨����Ҫ�����֤���ļ���ʽ�Ĺ��桷Ҫ��
���������ٴ�����
1.��Ʒ����Ҫ���鱨��
1.1��Ʒ����Ҫ��
ע��������Ӧ����ԭ�������������������ȶ���ǰ���£����ݲ�Ʒ���ơ�ǰ�����۵Ƚ�������ݹ��ұ�����ҵ�����й��������ϣ���ϲ�Ʒ�����ա�ҽ����е��Ʒ����Ҫ���дָ��ԭ��Ҫ���д��Ӧ���ٰ����Բ�Ʒ���Է����ʡ����Է����ʡ��ظ��ԣ�Ӧ����ͼ��Ũ�Ƚ��У����������Ŀ��Ҫ��Ӧ����Ҫԭ���ϼ���������Ҫ���������Ϊ��¼���ڼ���Ҫ�����ĺ�
��Ʒ����Ҫ��¼�У���Ҫԭ����Ҫ��Ӧ������������ߡ��ʿ��߰��������ǵĿ�ԭ�����������Դ��Ϣ������¡�������¡���������ԭ������ѧ��Դ����Ϣ��������ά��Ĥ�ĺ�ȡ�����С����Ĥ�ٶȼ��������������Ϣ����Ϊ����ԭ����Ӧ�����Ʊ��������巽��������Ҫ����Ϊ�ԭ����Ӧ����������Ƽ���Ʒ����������Ҫ����Ҫ��Ӧ��Ҫ��������������Ʊ�������ߺ��ʿ����Ʊ��ȸ�������̷�����������ʪ��Ҫ���ʿر�������Ũ��Ҫ��ȡ�
�������õĹ��ұ�����ҵ������Ʒ����Ҫ������Ҫ��Ӧ��������Ӧ��Ҫ��
1.2��Ʒ���鱨��
�����״�ע���Ʒ��ע���������ⶨ��Ʒ����Ҫ���Ӧ��������ͬ���β�Ʒ���м��顣�����Ѿ��й��Ҳο�Ʒ�ļ����Ŀ���ڼ��ʱӦ������Ӧ�Ĺ��Ҳο�Ʒ���У�����Ŀǰ�����Ҳο�Ʒ����Ŀ��������ҵӦ�����Լ����ʿ���ϵ���ṩ��Ӧ���ڲ��ο�Ʒ��
2.���������о�
ע��������Ӧ�����ڷ�������������ϵ�Ļ������������Լ��н������з��������о����ύ�����о�Ŀ�ġ�������ơ��о��������ɽ��ܱ����������ݡ�ͳ�Ʒ�����������۵���ϸ���ϡ��йط���������֤�ı�����ϢҲӦ���걨�������������֣���������ص㣨ʵ���ң����Լ�������š�������Դ�ȡ����������о����鷽���ɲο������Լ����������Լ�������������ע�����ָ��ԭ�������ٴ�ʵ���ұ���Э����ָ�ϣ�CLSI-EP���ļ��ȹ������й�������ϲ�Ʒ����������ָ���ļ����С����걨��Ʒ������ͬ�İ�װ����磬���͡����͡���Ɑ�͵ȣ�����Ҫ�Ը���װ�����з�������֤�����ڲ�ͬ����������Ӧ�ֱ��ύ��Ӧ�ķ��������������ϡ�
����ҩ�����ü���Լ��������ص�����·������ܽ����о���
2.1�����ȶ���
ijЩҩ�����������ù������д�л���������̣����Ӧ�������������ȶ��Ե��о���˵������ҩ���������е��ȶ��ԡ�ע��������Ӧ��ֿ���ʵ��ʹ�ù����������ɼ������估����ȸ����ε��������Բ�ͬ�����������ȶ��Էֱ�������۲��ύ�о����ϡ����ݰ�������ı������������ʱ�����������������漰���ȡ�
2.2���õ���������
�걨����Լ��������ͽ���Ϊ��Һ����Һ�ȣ�Ӧ�ܸ����ٴ�ҽʦ�Խ��ڻ���ҩ��ʹ����������жϣ��ҽ�����ҩ�����õij�ɸ��ÿ��������������Ӧ�ֱ���з�������������
2.3ȷ��
ȷ�����۷�������ʹ���ο�Ʒ�ļ��������ѧ�ȶԣ���ҵ�ɸ���ʵ�����ѡ��������������о���
2.3.1�ο�Ʒ�ļ��
�ο�ƷӦΪ�ɾ��й�����ֵ�ı�Ʒ��ο�Ʒ���ö��õ��������������������ٴ���������һ���������������������Ũ��������ҩ����Ҫ��л��������ã�����ṹ����������ã���
2.3.2����ѧ�ȶ�
����ѧ�ȶԷ�Ϊ��������ͬ���Լ��ıȶԺ���ο������ıȶԡ�
ѡ�������е�ͬ���Լ��뱻�����Լ����з���ѧ�ȶԣ���ѡͬ���Լ������Ӧ���ڻ���ڱ������Լ�����ޣ�ͨ���ȶ�˵���������Լ����������Լ�֮���һ���Գ̶ȡ��о�Ӧ����һ�����������Ժ�������������ע������������Լ�������������;������Խ��淴Ӧ��������
�ο������ıȶԣ�����Һ��ɫ��-������LC-MS��������ɫ��-������GC-MS������ЧҺ��ɫ����HPLC���ȣ�Ӧ���ݴ��⻯��������ѡ�����˵IJο��������о�Ӧʹ����ͳ��ѧ����������������Ӧ����������Ϊ��������������������������Ũ������Ӧ���Ǽ���ޡ����������Լ50%Ũ�ȣ������ٴ����ѻ�õĵ�ֵ�������ɲ������Ի��ʶԸ�ֵ��������ϡ�͵ķ�ʽ��ã�����ϡ����������˵����������Ӧ������֪�������Խ��淴Ӧ�Ե�����ҩ����л�����ȼ���Լ������˶�����������жԱ������⣬��Ӧ���Ƕ���Ҫ��л����Ϊ��ȵ�ԭ��ҩ���صĴ�л�����O6-����������Լ������ɶ�������ɴ���ȣ����вο������ıȶԡ�Ӧ�ص����Щ������������Ũ�Ƚ��п��졣
2.4���ܶ�
Ӧ�Ծ��ܶ�ָ�꣬���������ϵ���ȵ����۱���������Ҫ���ܶ��о�Ӧ�����ٴ�����������ȷʵ���Ի�ȡ�ٴ������ı�����ڲ�������ԭ���ͬʱ����ʹ�ñ��������ӵ����Ի����н����о���
�о�Ӧ�������С�ʱ�䡢�����ߡ������������ã����Լ����κ͵ص��Ӱ�쾫�ܶȵ���������ƺ����ľ��ܶ����鷽���������ۡ��趨�����ľ��ܶ��������ڣ����磺Ϊ������20��ļ�⣬���巽���ɲο�������������ļ����С�
���ھ��ܶ����۵��ٴ�����Ӧ���ٰ���3��ˮƽ�����������������ˮƽ���������л�ǿ�����������������ݲ�Ʒ�����趨�ʵ��ľ��ܶ�Ҫ��
2.4.1����������������Ϊ��Ũ�ȣ����Է�����ӦΪ100%��n��20����
2.4.2�����ˮƽ�����������ˮƽ�������Է�����Ӧ��95%��n��20����
2.4.3��/ǿ����������������Ũ�ȳ��жȵ�ǿ���ԣ����Լ����Ϊ100%��CV��15%��n��20���������������ɫ��һ��
2.5�����
2.5.1�����ȷ��
Ŀǰ������ҩ�����õĶ��Լ���Լ��ɲο��ҹ���ؼ���ָ�ϼ������������ú;�����������֣�Substance Abuse and Mental Health Services Administration��SAMHSA��ȷ���ļ���ޣ���ɿ���Ƭ�����������ȣ�����������Ƽ�����ļ���Լ���ע�������˿ɸ������ҩ������л�������ȷ�������ļ���ޡ����Ʒ���������ҩ����Ҫ��л���P��ṹ���������ͬʱ������м����ȷ����
2.5.2�������֤
ע���걨��ƷӦ�����ڼ����ȥ25%��Ũ�ȡ�����ͼ������25%��Ũ���϶Ծ���ͳ��ѧ�����������ٴ��������з�����֤������ȷʵ���Ի�ȡ�ٴ������ı�����ڲ�������ԭ���ͬʱ����ͨ������֪����ҩ�����м���Ŀ��ҩ��ķ������������Ʊ�������Ũ�ȿ�ͨ������ɫ��-������GC-MS�����������õĵ�Ч��������ȷ����������Ҫ����ʵ�����Ũ�ȷ�Χ���������ں��ڼ����50%��75%��100%��Ũ��ˮƽ����������Ӧ���顣
�������߲���������Լ��Ľ���ж��������ǽӽ������Ũ�������Ľ���ж����ܲ�����Ա���������ж������Լ��Լ����������ε�Ӱ�졣��ˣ�������о������ɶ���������Ա���������ˣ�ʹ�ö�����εIJ�Ʒ�������������������顣����о��μ���Ա������������������䣬����������Ž����ڱΣ��Ա�����ֽ���ж��ϵ�ƫ�롣
2.6�������о�
2.6.1���淴Ӧ��֤
�������ͬ��ҩ���е�����ҩ��/�������ҩ���л�����ܷ������淴Ӧ�Ľṹ��������н��淴Ӧ��֤��
��ʹ����ҩ����������ҩ�ﻯ����ķ����Ʊ��������о���������Ũ��ˮƽӦ�����������Ԥ��ˮƽ�൱�����ij����������Ϊ���ԣ����Ըû�������������ϡ�ͣ�ֱ��������Խ����
�ܹ���ñ�Ʒ����Ҫ��л���P�ṹ������Ӧ�����������ȣ���ijЩ��֪��Ҫ��л�����Ʒ���ɵã��ɶ������ٴ�������������ɫ��-���ף�GC-MS������ͨ������ͼ�ƶ���ṹ��֤�����������еĴ��ڡ�
2.6.2��������
DZ�ڵĸ���������Ҫ�������������ռ���л�ij���ҩ������ʣ���Դ�����ʻ����������仯��������Щ���ʼ����������Ƿ��Լ�������Ӱ�졣������ҩ�����Ӧ�������ô������Ǵ���ҩ�����Ѫ������ҩ�����ҩ�������ҩ������غͽ�����ʹҩ��ȣ�
Ӧʹ��ҽѧ���ˮƽ�ĸ�����Ũ�Ƚ�����֤�����⣬�ཨ��ע�����������ÿ�ָ������ʵ�DZ�����Ũ�ȣ���������������������ۡ����ڳ���ҩ��������飬���������Ӧҩ��ҩ������ѧ�о�ȷ��������ҩ��Ũ�ȼ���θ�ҩ���Ũ��ˮƽ������Ӧҩ���Ʊ������������и�����֤����Դ�����ʣ���������Դ�Ը�����ĸ���Ӧѡ����Ӧ��ֵ������������顣
�����ڼ������ˮƽ��ÿ�ָ������ʵ����Ժ����Ը���Ӱ��������ۡ��ֱ��������ٴ������м������Ũ�ȵĸ���ҩ��/�����P��Դ�����ʣ�һ��Ϊ��֪��õ����Խ�������Ũ��Ŀ��ҩ��������Ը���Ӱ�죩����һ��Ϊ��֪��õ����Խ�������Ũ��Ŀ��ҩ��������Ը���Ӱ�죩������۲췢��Ԥ�ڽ�����ֱ仯������Ը����������ϡ�ͣ�ֱ������������Ӱ�졣
�������������仯�Լ���Ӱ�죬�ɶ���������Ũ�ȵ��������е������Է�ӳ���ڿ��ܴ��ڵĸ������������仯����ɽ���Һ���������pHֵ��Χ��Ϊ3��9���۲첻ͬpH�����¼������Ԥ�ڽ���Ƿ���ڱ仯�����ڱ����Լ�����ж�������ɫ���۹۲죬����Թ��±�ɫ���ʵ�Ӱ��������ۣ�����Ѫ�쵰�ס����쵰�������˹�����Ȼ�γɵ�ʳ��ɫ�ء�ҩ��ȣ���Һ���Ӧ�ֱ�Կ��ܶԲ�Ʒ���ܲ���Ӱ�����Դ�����ʣ�ճ���ס��ܾ�ø��������IgA�ȣ�����Դ�����ʣ���Һ���ڴ̼����������ء�������ʹҩ��ά���ء����ϡ����ࡢ����ˮ�ȣ���Ӱ����п��졣
2.7��Ӧ��ϵ
2.7.1��Ӧ����ȷ����ע��������Ӧ���Ƿ�Ӧʱ�䡢�ж�ʱ�䡢��Ӧ�¶ȵ������Բ�Ʒ���ܵ�Ӱ�죬ͨ������ȷ�����������������ϡ�
2.7.2��Ӧ��ϵ����Ʒ������ʽ��������ȷ����ͨ������ȷ����ѵļ�����ʽ����������
3.�ȶ����о�
�ȶ����о�������Ҫ����ʵʱ�ȶ��ԣ���Ч�ڣ��������ȶ����Լ������ȶ��Ե��о���ע�������˿ɸ���ʵ����Ҫѡ��������ȶ����о��������ȶ����о�����Ӧ�����о�������ȷ�����ݡ������ʵʩ��������ϸ���о������Լ����ۡ�����ʵʱ�ȶ����о���Ӧ�ṩ��������������ʵ�ʴ��������±�������Ʒ��Ч�ں���о����ϡ�
4.�����ж�ֵ�о�
ҩ�����ü���Լ�����/���ԵȽ���жϵ������ж�ֵ��cut-off,CO����ʹ���Լ������ļ���ޣ����Ʒ���������ҩ����Ҫ��л���P��ṹ�������Ӧ�Դ�л���P�ṹ���������ͼ��Ũ��ˮƽ�����о���
Ӧ�����������͵��ٴ��������������ж�ֵ����֤�����ύ������Դ��������ȷ�����ݡ���֤���鷽����ͳ��ѧ�������о����ݵȡ�
5.��������
5.1��Ҫԭ�����о�����
���߲����������ҩ�����ü���Լ�һ����þ�����ԭ����Ӧ�ṩ��Ҫԭ�����������ϰ����Ŀ�ԭ������굥��¡�����Լ���ҵ�ο�Ʒ�ȵ�ѡ������Դ���Ʊ����̡��������ȵ�����о����ϡ�����Ҫԭ����Ϊ��ҵ���ƣ�Ӧ�ṩ����ϸ�Ʊ����̣�����Ҫԭ����Դ�����Ӧ�ṩ�����ϰ�����ѡ���ԭ���ϵ����ݼ��Ա�ɸѡ�������ϡ��������ṩ���������������춨���棬�Լ���ԭ�ϵ�����������������ϣ���Ӧ��Ӧ�̶����������������ע��������Ӧ�Ը���Ҫԭ���Ͼ���ȷ�������Ʊ���
5.1.1��ԭ����
���ڽ�����ǡ�����������ά��Ĥ�������Ʊ�����ߡ��ʿ��ߵĿ�ԭ����ȣ��翹ԭ������굥��¡����ȣ�Ӧ�ύ��ԭ/������Դ���Ʊ���ɸѡ������������������ۡ�����Ũ�ȡ����ȡ���������Ч�ۡ�����������ȣ�����ϸ�������ϡ�
5.1.2����
������ԭ�����⣬��Ʒ�а���������ԭ���ϣ��罺���������ά��Ĥ��������ά��Ĥ�ȣ���Ӧ����ѡ����֤�����ύ������ϡ���ȷ��Ӧ�̺��������Ʊ���
5.1.3��ҵ�ο�Ʒ
��Ʒ����ҵ�ο�Ʒһ��������Բο�Ʒ�����Բο�Ʒ������ο�Ʒ���ظ��Բο�Ʒ��Ӧ���ݲ�Ʒ������֤��ʵ����Ҫ������ҵ�ο�Ʒ��
Ӧ�ύ��ҵ�ο�Ʒ��ԭ����Դ��ѡ���Ʊ��������Լ�Ũ��ȷ�Ϸ������Լ��������֤���ϡ�ʹ����֪�������Ʊ���ҵ�ο�Ʒʱ������Ӧ�������ٴ��������ƵĻ��ʲ�˵�����ʵľ������ƹ��̣�����Һ�������ʣ�����ȷ�˹���Һ���Ʊ�������Ӧ��ע�������ñ����ʵijɷ֣���Ϊ�����Ρ������εȡ�Ӧ�������Ʋο�Ʒ�����м���ο�Ʒ��������ľ���Ũ�ȣ�����Ӧ�����ﵥ�������Ũ����Ϊ�ο�Ũ�ȡ���ҵ�ο�Ʒ�����ý������£�
5.1.3.1���Բο�Ʒ
��ҵ���Բο�Ʒ����Ӧ��������������������Ҫ��л���P�ṹ�����������������ã���
5.1.3.2���Բο�Ʒ
��ҵ���Բο�Ʒ���ó�Ӧ�����������������⣬��Ӧ����������������ŵĽṹ�������������ҩ��Ⱥ�ҩ������
5.1.3.3����ο�Ʒ
������ϵ��ϡ������������Ӧ���������ˮƽ��
5.1.3.4�ظ��Բο�Ʒ
��������ߡ�������Ũ�ȵ�����������һ��Ũ��ӦΪ���������Ũ�ȡ�
5.2�������������
5.2.1��Ʒ������Ӧԭ�����ܡ�
5.2.2��Ҫ�������ս��ܣ���������ͼ��ʽ��ʾ������Ҫ˵����Ҫ�������յ�ȷ�����ݡ�
5.2.3�������塢��ɫϵͳ�Ľ��ܼ�ɸѡ�����ʷ�Ӧ�������о���
5.2.4����/��ǹ����о���ע��������Ӧ���������/���Һ����Ũ�ȡ�ʱ�䡢������ָ��Բ�Ʒ���ܵ�Ӱ�죬ͨ������ȷ������ָ��������ϡ�
���ģ��ٴ���������
���롶�����ٴ�������������Լ�Ŀ¼����ҩ�����ü���Լ�����ͨ���뾳��ͬ�������в�Ʒ�ķ���ѧ�ȶԽ����ٴ����ۣ����۷����ɲ��ա������ٴ��������������Լ��ٴ����ۼ���ָ��ԭ��������ݣ���������������Ӧ����������ҩ�����õĽ��淴Ӧ���������ó���ҩ���DZ�ڸ������������ڰ������ְ�װ����磬���͡����͡���Ɑ�͵ȣ��IJ�Ʒ����ͬ��װ�����ڷ�Ӧ��ϵ�IJ��죬�ٴ�����Ӧ���Dz�Ʒ���а�װ������л���ͳ�Ʒ�����ͬʱ�Բ�ͬ��װ�����зֲ�ͳ�ơ�
�����뾳��ͬ�������в�Ʒ�ķ���ѧ�ȶ������ṩ��������Ϣ��Ϊ���ޣ��ҿ���Դ���ҩ��/������ķ�Ӧ�Բ�ͬ���Լ����ǵ����淴Ӧ�����ء�ͬʱ����Ӧѡ���ⷶΧ��һ��������60�����类�����Լ�����ͬ�������в�Ʒ����Ӧ���������������������ھ߱��й��ϸ����������Ͽ�ίԱ�ᣨCNAS���Ͽɵ�ʵ���ң���ο���������Һ��ɫ��-����������ɫ��-��������ЧҺ��ɫ���ȣ����бȶ����顣
��ο������ıȶ�Ӧ����������Ϊ��������������������������Ũ������Ӧ���Ǽ���ޡ����������Լ50%Ũ�ȣ������ٴ����ѻ�õĵ�ֵ�������ɲ������Ի��ʶԸ�ֵ��������ϡ�͵ķ�ʽ��ã���Ӧ��ϡ����������˵����
Ӧ�Բο������ķ���ѡ����ѧ������������ϸ������������ɫ��-������GC-MS��Ӧ��ϸ����������������������ɫ������������������������Ӳ��ṩ����ɫ��ͼ������ͼ����˵����ȷ���ļ���������з���ѧ����ľ��������緽��ר���ԡ������ȡ����ԡ����ܶȡ������ʵȡ�
����ijЩҩ���������д��ڷֽ��Ӧ���̣����Ϊ��֤�Ա���������ȷ��Ӧ����ͬ�����вο������ͱ������Լ��ļ�⣬�������ּ�ⷽ��֮�������ij�ʱ����á�
���Ŀǰ����ͬ���Ʒ���е����Ͷ�Ʒ����Լ������������������͵Ķ�Ʒ����Լ���Ӧͨ���ٴ�����·�������ٴ����ۡ��ٴ�����Ӧ���ϡ���������Լ�ע���뱸�������취����ҽ����е�ٴ��������������淶���͡���������Լ��ٴ����鼼��ָ��ԭ��Ҫ������ط��桢�ļ��и��£��ٴ�����Ӧ���ϸ��º��Ҫ�������˵�������Ʒ�ٴ�������Ӧ��ע���ص����⡣
1.�ٴ��������
Ӧѡ��߱���Ӧ�����Ұ��չ涨������ҽ����е�ٴ����������չ�ٴ����顣���ڸò�Ʒ��������ʹ��Ŀ�ģ��������Ѿ�������ר��ҽԺ���䶾���ĵ�������ƻ�����չ�ٴ��о����ٴ������������Ӧ������3�ң��Ҿ�רҵ���ƣ�ʵ�������ԱӦ���㹻��ʱ����Ϥ���ϵͳ�ĸ����ڣ���Ϥ���۷����������������У�������������Լ��ͶԱȷ���/�Լ���Ӧ������Ч�����������£�����ȱ�֤�������ݵ�ȷ�Լ����ظ��ԡ�
2.�ٴ����鷽��
2.1���Ͷ�Ʒ����Լ��ٴ����鷽��
�����˿ɲ���������������Լ�����Ӧ�ο�������������ɫ��-��������ЧҺ��ɫ���ȣ������ٴ��ȶԣ�����������������Լ����ٴ��ο������������һ���ԡ�����ٴ��ο�����Ӧ��ȷ�������ж�ֵȷ���������ṩ������ݡ�
�ٴ����������и��һ���ʹ�õ��ٴ��ο�����Ӧһ�¡�Ӧ���ٴ��ο�����������ϸ�Ľ��ܣ����ύ������֤���ݣ�֤���ٴ��ο�������������������Լ��Ŀɱ��ԡ����ٴ��ο�����ί������������ɣ���Ӧ�ύ���ٴ��������ί�е���������/ʵ���ҿ�չ�������ķ����ͬ/Э�飬�ύ��ػ������ʣ�ʵ����Ӧ�߱��й��ϸ����������Ͽ�ίԱ�ἴCNAS���ʵȣ���ѡ�����ݡ�
2.2 ���������������͵Ķ�Ʒ����Լ�
�����Ʒ�ٴ������ѡ������Ӧ�ο�������������ɫ��-��������ЧҺ��ɫ���ȣ������ٴ��ȶԣ�������������Լ����ٴ��ο���������ӦΪͬһ������������Ҫ�����2.1��
���⣬������������Ѿ����ϵ������������еIJ�Ʒ���ٴ����黹Ӧѡ�ֲ������������еIJ�Ʒ��������������Լ����й����������������������͵ıȶ��о���
3.�ٴ�����������Ⱥ��ѡ��
�ٴ������������ȺӦ���Բ�Ʒ��Ԥ��������Ⱥ���ò�Ʒ��������ȺΪҩ��������Ⱥ�����������ѡ��Ŀ����Ⱥ�⣬��Ӧѡ������ҩ�����õĽ��淴Ӧ���������ó���ҩ���DZ�ڸ��Ų����������ٴ�������ԴӦ���ݣ������ٴ�����Ӧ���벿��������������
4.�ٴ�������������
�����Ʒ�����漰���������Ͱ�����Һ����Һ�ȡ��ٴ������IJɼ����鰴�����ʵ���Ҽ�⼼������ִ�С�
�ٴ��о���ԱӦע��ijЩҩ�����ٴ������еĺ�������ʱ������ƶ����٣�����Ʒ�����������������ϣ�Ӧ�����ܵ������ڿ����Լ���Ա��Լ�/�ο����������������ʱ�䣬�����ɴ˴�����������ƫ�
5.�ٴ�����������
�ٴ���������������������������Ӧ�ֱ�����ͳ��ѧҪ��������ٴ��ο������ĶԱ����飬�ɲ���Ŀ��ֵ����ʽ�ֱ����������Ժ���������������
���������㹫ʽ���£�
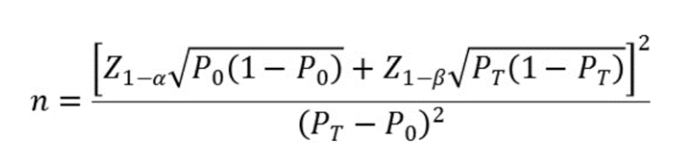
��ʽ�У�nΪ��������Z1-����Z1-��Ϊ������ˮƽ�Ͱ��նȵı���̬�ֲ��ķ���λ��P0Ϊ����ָ����ٴ��ɽ��ܱ���PTΪ������������Լ�����ָ��Ԥ��ֵ��
�������Է����ʺ����Է����ʵ��ٴ��ɽ��ܱ���P0�����鲻����95%���ٴ��������У��������ָ���95%������������Ӧ������Ԥ����ٴ��ɽ��ܱ���������ָ��P�ӽ�100%ʱ���������������㷽�����ܲ����ã�Ӧ����ѡ��������˵ķ������������������ͳ��ѧ�������羫ȷ���ʷ��ȡ�
������������������͵Ķ�Ʒ����Լ��������ڹ��ϵ��������͵Ķ�Ʒ����Լ�����ͬԴ�ȶ����飬��Ӧ����һ�����������Լ����Բ������ɲ��ó������ȵĹ�ʽ�������������㡣
6.�ٴ���������ͳ�Ʒ���
�ٴ�������һ�����ĸ������ʽ�����ܽᣬ���ݴ˼���������������Լ��������Ⱥ�����ȣ�����Աȷ���������/���Է����ʼ���95%�������䡣
�ٴ����鱨����Ӧ�����������ߵĻ���������з������������������䡢�Ա�ķֲ�������Լ��ٴ���ϱ����ȡ��ر�Ӧ����������������۵ĸ��������߽��й�����ܣ�ȷ�������������нϺõĴ����ԡ��ٴ����������漰��ͬ�������ͣ�Ӧ���ÿ���������ͷֱ����ͳ�Ʒ�����
�ٴ����������в�һ�½����Ӧ��ϲ�����ҩ������ʷ��ҩ��ʹ������ٵ���Ϣ���г�ֵķ������ٴ�������Ӧ�ܹ�֤����Ʒ�ٴ����������ٴ�Ҫ��
7.�����ٴ��������ݵ��Ͽ�
�����ٴ���������Ӧ���ϡ�����ҽ����е�����ٴ��������ݼ���ָ��ԭ�͡�ʹ����������Լ������ٴ��������ݵ�ע�����ָ��ԭ�����Ҫ���ύ�������ٴ����鷽����������������������Լ��������������й�������Ⱥ����֤���ϡ��������ٴ�����������������ĶԱ����Ϻ��ٴ���������������������ٴ�������Ӱ�����֤���ϡ�
ע��������Ӧ���������ٴ����鼼������Ҫ����֤�����ٴ��������ݵij���ԡ�
8.�ٴ�֤�ݵ���ʽҪ��
������Ӧ���ա���������Լ�ע���뱸�������취�������ڹ�����������Լ�ע���걨����Ҫ�����֤���ļ���ʽ�Ĺ��桷�ȷ����ļ�Ҫ���ύ�������������������ٴ����鷽�����ٴ�����С�ᡢ�ٴ����鱨���Լ��ٴ��������ݿ⡣
���壩��Ʒ˵�����ͱ�ǩ����
˵��������˲�ƷԤ����;����������������������Ľ����Լ����ע���������Ҫ��Ϣ����ָ��ʹ����Ա��ȷ��������Լ�������������ҽѧ���͵���Ҫ���ݣ�Ҳ����������Լ�ע���걨����Ҫ�ļ�֮һ��ҩ�����ü���Լ�ͨ������ҩ�����õij�ɸ��⣬������Ԥ����;�������ԣ�˵�����ж�Ԥ����;��ȷ������������Ҫ����ɼ����������Ľ��͡����鷽���ľ����ԡ�ע����������ݵĽ�����Ϊ��Ҫ��������ʹ������ȷʹ���Լ�����ȷ��������
��Ʒ˵������������ݾ�Ӧ��ע���������ύ��ע���걨�����е�����о��������һ�£���ijЩ���������Բο����ף���Ӧ�Թ淶��ʽ�Դ����ݽ��б�ע�������������ο����������Ϣ�������ط���Ҫ��ҩ�����ü���Լ������ԣ������˵������ص���������ϸ˵����
1.����Ʒ���ơ�
��Ʒ��ͨ�����ư��ա���������Լ�ע���뱸�������취����ͨ�����Ƶ�����ԭ��ӦΪ�����������ƣ�����Լ��У�����ѧ�����磺��ȼ���Լ��У����������Ϊ�������Լ��У�������Ϊ�����������ƣ��ֱ��г��������ϼ���Լ��У�����ѧ�����磺��ȡ��Ȱ�ͪ���ϼ���Լ��У����������Ϊ�������Լ��У��Ұ�װ�����в�ͬ�������ʽ��������Ϊ�����Ʒ����Լ��У�����ѧ�������Լ�������Һ������������Ϊ�����������ƣ���Һ����Լ��У�����ѧ�����磺�����Һ����Լ��У��������
2.����װ���
�Բ�Ʒ��װ��������Ӧ�����Լ�����ͬ�����ʽ�������ͣ����͡����͡���Ɑ�͵ȣ��Լ�ÿ��װ���������˷ݣ����磺�����/�Ȱ�ͪ����Լ������ͣ�40�˷�/�У����ͣ�50�˷�/�У����/�Ȱ�ͪ/��������������Լ������ͣ�40�˷�/�У����ͣ�50�˷�/�У����ͣ�5�˷�/�С���
3.��Ԥ����;��Ӧ���ٰ������¼��������ݣ�
3.1��Ԥ����;�ľ�������Ӧ�����������ݣ��ò�Ʒ���ڶ��Լ�⣬˵�����õ��������ͣ���ȷ������ͨ��������������������ա�����ҩƷƷ��Ŀ¼����/����ҩƷƷ��Ŀ¼����ҩƷͨ�������ұ�����ӦΪҩ�����á�ҩ����Ҫ��л���P��ṹ�����˵��������ļ���ޣ���ǿ�����ڡ�������ij�ɸ��⡣�磺�ò�Ʒ���ڶ��Լ��������Һ����ͼ��Ũ��Ϊ50 ng/mL�����������ᣬ��������������ij�ɸ��⡣
3.2������ҩ�����������������ʽ�����ӽṹ����ṹ�������Ҫ��л;����ҩ������ѧ��������˥�ڣ�����Ҫ��л���P��ṹ���������ҩ���ҩ�����ã����ú��µ����弰������֡�
3.3���������ȷ�Ϸ�����
Ԥ����;��Ӧ�������Լ�������ҩ�����õij�ɸ��⣬��Ҫ��һ��ȷ�ϼ����۵�����Ӧ���������ȼ������Ը��ߵļ�ⷽ�����С��磺Һ��ɫ��-������LC-MS��������ɫ��-����(GC-MS)����ЧҺ��ɫ����HPLC���ȡ�
4.����Ҫ��ɳɷ֡�
4.1Ӧ�����Լ���/��/���ṹ��ɡ�����ߡ��ʿ��߰��������ǵĿ�ԭ���������ѧ��Դ��Ϣ���翹ԭ��������¡�������¡���塢����Ķ���Դ�Ե���Ϣ��
4.2Ӧ˵��������Ҫ�����Լ���δ�ṩ����Ҫ���ϣ����ṩע��֤��/�����ŵ���Ϣ��
5.��������������Ч�ڡ�
������������Ч�ڰ����Լ��е�Ч���ȶ��ԡ������ȶ��ԡ������ȶ��Ե��й��Լ��������Ҫ��Ϣ�����б�Ҫ��Ӧע���Լ�����仯�����ʱ�������������ؾ�ʾ��
Ӧע������������Լ��Ի����¶ȼ�ʪ�ȵ�Ҫ���������ȡ�
6.������Ҫ��
Ӧ��ȷ�������͡��ɼ������������ɼ���ע�������������Ӧʹ�õ��ռ����������������ȶ��ԵĴ��桢��������������ʱ�䣻��������ʹ�õ��������ͣ�����������������������Һ�����Ƿ���Ҫ���ġ����˵ȣ�˵�������ɼ������治��������ɵ�Ӱ�졢�����п��ܺ����������ʣ�Ӧ��ȷ����ɵ�Ӱ�졣
��������Һ�����ļ�⣬Ӧ��ϸ�涨��Һ�����IJɼ�����������������λ��ͣ��ʱ��ȡ��÷���Ӧ�������õ��ظ��ԣ����ܱ�֤�ɼ����Ļ����㶨����Һ����Ӧע����ʾ�����ռ�ǰӦ������ˮ����ʳ�����̡��������ǵȿ��ܸ��ż����������Ϊ��
7.�����鷽����
������������ͼʾ��ʽ��ʾ��ȷ�ļ����������������ע���������ͼʾ��ʾ����ȷ�IJ��������ȡ��ر�ע��Ӧǿ�������¶ȼ�ʪ����������ȡ�����ʱ�䡣
8.�������ж�ֵ��
Ӧ˵���������ڴ������������пɼ������ͼ��Ũ�ȡ������Լ����ܹ�����Ŀ��ҩ��/����������������ж�ֵ���п�ѧ���������Ʒ�����ﻹ����ҩ����Ҫ��л���P��ṹ������������ж�ֵ������Ӧ����Ϊ�ܹ����ij�����и���ijŨ�ȵ�ҩ���/���൱����ҩ����Ҫ��л����/�ṹ�����
9.���������Ľ��͡�
������ȷ�ü���Լ��������;��Χ���ܹ������ҩ�ҩ��ԭ�ͼ����л���ͬ��ҩ��/�ṹ������ȣ������ܿ������ҩ�������Ե�ԭ��˵��ҩ��ת��ȡ���ڶ������أ�����ҩ��ʹ��Ƶ�ʡ���ҩ������л�ʺ�����֬�������ȡ����ҩ���ҩ������ѧ��ҩ�����ã����ܴ���ҩ�������ڿ��ܴ��ڵ�ʱ���Լ����ܳ������õ�ʱ�䣬���������ɼ�ʱ���Լ������Ӱ�죬�����ڽ���Ľ��ͺ��жϡ�
������������ͼʾ��ʽ��ʾ���п��ܳ��ֵļ�����������ԡ����ԡ���Ч�ȣ���������Ľ��ͣ������۲쵽�ض����֮��Ӧ����ȡ�Ĵ������������ʿ���δ���֣���Ч��������������塢ԭ�������Ӧ��ȡ����һ������������������������Ч��Ӧ��ȡ�Ĵ�ʩ���Ƿ���Ҫʹ�������Ը��ߵ�ȷ�Ϸ����ȡ�����ҩ��֮������Ҫһ��ʱ��ҩ��Ż��������г��֣���ҩ���������д��ڵ�ʱ�����ޣ���������ɼ����������������ܻ�����Խ��������˵���������������ԣ���һ������δ����ҩ�������������û�з�������ҩ�����ܷ�����ҩ���δ�������Ӧ��������������Ҫ�������¼��������������ȷ�ϣ��绳��ҩ������ʱ��Ӧ��ѡ����һʱ���ٴμ�⣬�����������ͬ����ҩ����м�⡣
10.�����鷽���ľ����ԡ�
10.1ǿ�����Լ�ֻ���ڳ�ɸʹ�ã������������Ϊȷ��ҩ�����õ����ݡ��Խ����ȷ�ϱ���ʹ�������Ⱥ������Ը��ߵIJο�����������
10.2�����÷������ܵ��¼����Լ������Խ����ԭ��
��ɼ����Խ����ԭ�������������ҩ��Ũ�ȵ��ڼ���Լ�����ޣ���Ũ���������������������ȷ�����Ӱ������������أ������估���治��ʹ�Լ�ʧЧ�ȡ�
��ɼ����Խ����ԭ�����������ijЩ����ҩ���ʳ��ijЩʳ�����£����������
10.3����Ӱ���Ʒ���ܵ����ء�
11.����Ʒ����ָ�
��Ʒ����ָ�������Ӧ�����������������һ�£�ע�������˳����ṩ�����������ݵ�ժҪ֮�⣬��Ӧ������������Ӧ�����鷽����������ơ��ص㡢��Ա�����������ȣ����м�Ҫ�������������ݰ�����
11.1��Ӧ���Ҳο�Ʒ�ķ�������������ã���
11.2����ޡ������Ũ�ȸ�����������֤�����
11.3���ܶȵ��о������
11.4������������
���淴Ӧ��Ӧ�г����п��ܲ������淴Ӧ��ҩ��/�����P����ͼ��Ũ�ȣ����о���֤���淴Ӧ�Ե�ҩ��/�����P����֤�����Ũ�ȡ�
�������ʣ������������յij���ҩ������ʡ���Դ�����ʻ����������仯�Լ��ĸ�����֤�����
11.5�ٴ����۽�����ܽᡣ
12.��ע�����
12.1�й��Լ�����ע������籣�桢��������������Ҫ��ȡ�
12.2�й��������������ע������й����������ܵ���Ⱦ���µĴ�����������Һ�к���Ư���������Լ��������Ƿ��Ӱ����Խ���ȡ�
12.3��֤�������ȷ�ж���ע��������ȡ�����ʱ�䡢���������Լ����ж�ʱ������ɫ��dz���ж������Ӱ�졣
12.4���ﰲȫ�Ծ��棺�Ӵ������ٴ���������������һ����ʹ����Ʒ�Ȳ���Ӧ����ΪDZ�ڴ�Ⱦ����д��������Ҳ��÷��Ϸ����Ԥ����ʩ��
12.5��������Ӱ���������Ⱥ������Ե�ע�����
�ġ��ο�����
[1]�����г��ල�����ܾ�.��������Լ�ע���뱸�������취�������г��ල�����ܾ����48��[Z].
[2]�����г��ල�����ܾ�.��������Լ��ٴ����鼼��ָ��ԭ����ҩƷ�ල������ͨ��2021��72��[Z].
[3]����ҩƷ�ල������.���Լ����������Լ�������������ע�����ָ��ԭ����ҩƷ�ල������ҽ����е������������ͨ��2022���36��[Z].
[4]����ҩƷ�ල������.�����ٴ�������������Լ�Ŀ¼������ҩƷ�ල������ͨ��2021���70��[Z]..
[5]����ҩƷ�ල������.�����ٴ��������������Լ��ٴ����ۼ���ָ��ԭ����ҩƷ�ල������ͨ��2021���74��[Z].
[6]Mandatory Guidelines for Federal Workplace Drug Testing Programs: Federal Register. ,2008[Z].
[7]Premarket Submission and Labeling Recommendations for Drugs of Abuse Screening Tests. Draft Guidance for Industry and FDA Staff[Z].